 Chugai Pharmaceutical – mRNA Display Platform & LUNA18 (pan-KRAS Inhibitor)")
안녕하세요 보스턴 임박사입니다.
일본의 제약회사 중 하나인 Chugai Pharmaceutical Co. Ltd. (中外製薬株式会社)는 2002년부터 Roche의 계열사로 되어 있는 회사로 신약개발을 하는 회사입니다. Chugai가 최근 mRNA Display Platform을 통해서 Macrocyclic Peptides 를 통한 신약개발을 중점적으로 하고 있는데 그에 대해 얘기를 하려고 합니다.
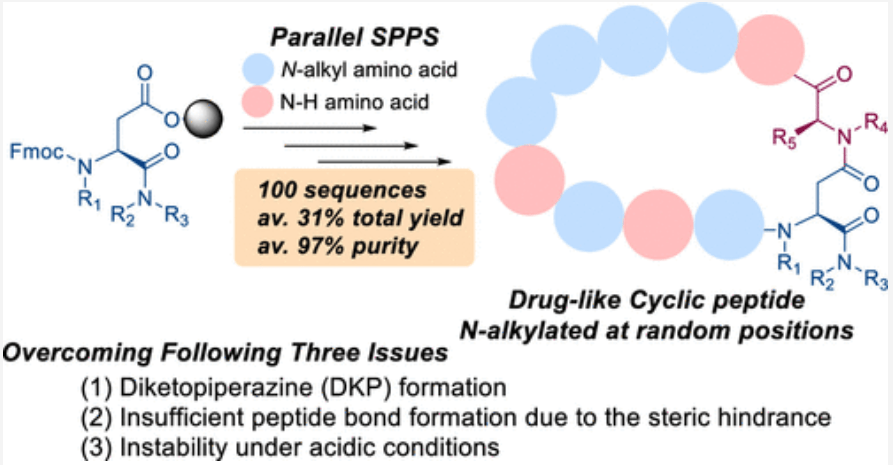
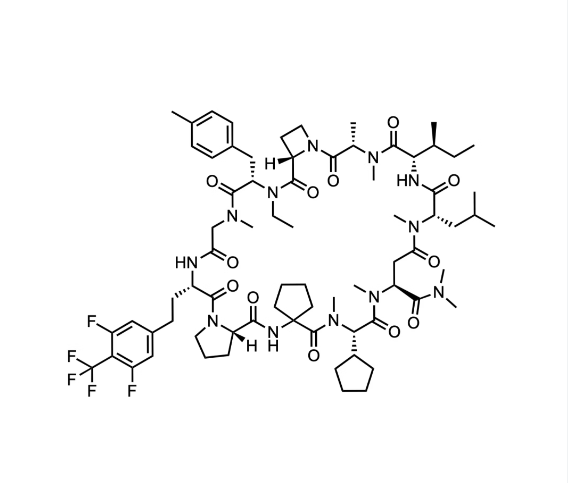
We report a versatile and durable method for synthesizing highly N-alkylated drug-like cyclic peptides. This is the first reported method for synthesizing such peptides in parallel with a high success rate and acceptable purity that does not require optimizations for a particular sequence. We set up each reaction condition by overcoming the following issues: (1) diketopiperazine (DKP) formation, (2) insufficient peptide bond formation due to the steric hindrance of the N-alkylated amino acid, and (3) instability of highly N-alkylated peptides under acidic conditions. Using this newly established method, we successfully synthesized thousands of cyclic peptides to explore the scope of this modality in drug discovery. We here demonstrate the syntheses of a hundred representative examples, including our first clinical N-alkyl-rich cyclic peptide (LUNA18) that inhibits an intracellular tough target (RAS), in 31% total yield and 97% purity on average after 23 or 24 reaction steps.
최근에 Journal of American Chemical Society에 LUNA18의 신약개발에 대한 논문을 게재했습니다.
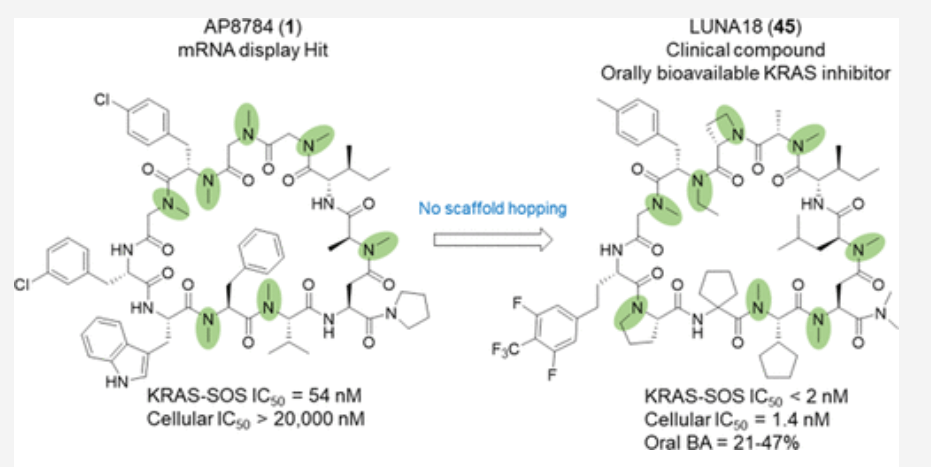
Cyclic peptides as a therapeutic modality are attracting a lot of attention due to their potential for oral absorption and accessibility to intracellular tough targets. Here, starting with a drug-like hit discovered using an mRNA display library, we describe a chemical optimization that led to the orally available clinical compound known as LUNA18, an 11-mer cyclic peptide inhibitor for the intracellular tough target RAS. The key findings are as follows: (i) two peptide side chains were identified that each increase RAS affinity over 10-fold; (ii) physico-chemical properties (PCP) including Clog P can be adjusted by side-chain modification to increase membrane permeability; (iii) restriction of cyclic peptide conformation works effectively to adjust PCP and improve bio-activity; (iv) cellular efficacy was observed in peptides with a permeability of around 0.4 × 10–6 cm/s or more in a Caco-2 permeability assay; and (v) while keeping the cyclic peptide’s main-chain conformation, we found one example where the RAS protein structure was changed dramatically through induced-fit to our peptide side chain. This study demonstrates how the chemical optimization of bio-active peptides can be achieved without scaffold hopping, much like the processes for small molecule drug discovery that are guided by Lipinski’s rule of five. Our approach provides a versatile new strategy for generating peptide drugs starting from drug-like hits.
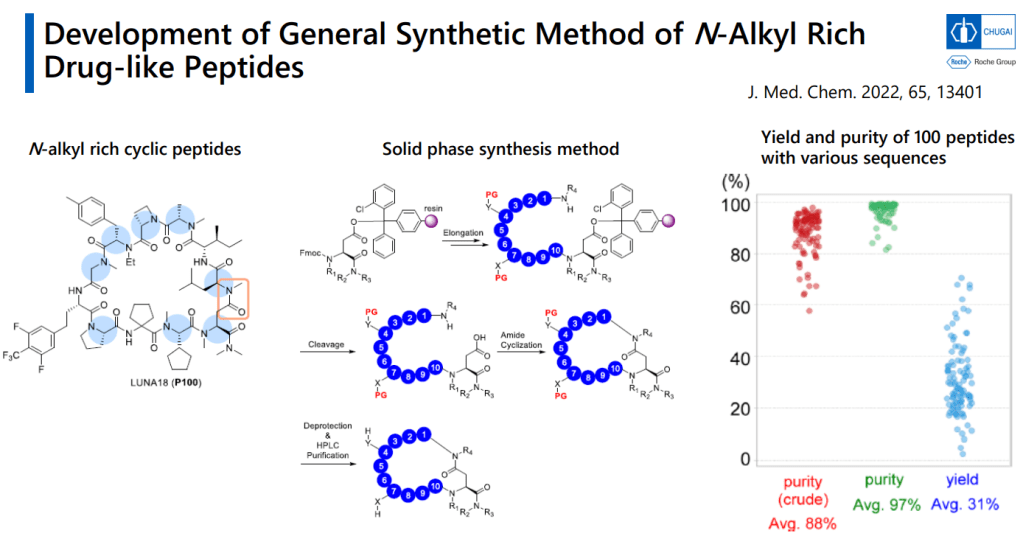
Journal of Medicinal Chemistry 2022년 논문에서 N-alkyl Rich Drug-like peptide synthesis를 개발해서 한 연구원 당 1년에 500개의 Peptides를 만들 수 있도록 했습니다.

Solid-phase synthesizer로 만드는 방법입니다. Cyclization을 먼저 한 이후에 Protecting group deprotection을 합니다.
최근에 Chugai R&D Day에서 발표한 자료에도 LUNA18에 대한 내용이 있습니다.
Chugai 제약은 Small molecule과 Antibody 신약개발은 오랜 기간 내공이 있는데 최근에 “intracellular Transferring Peptides” 분야에 Positioning을 한다는 전략을 수행하는 중에 있습니다.
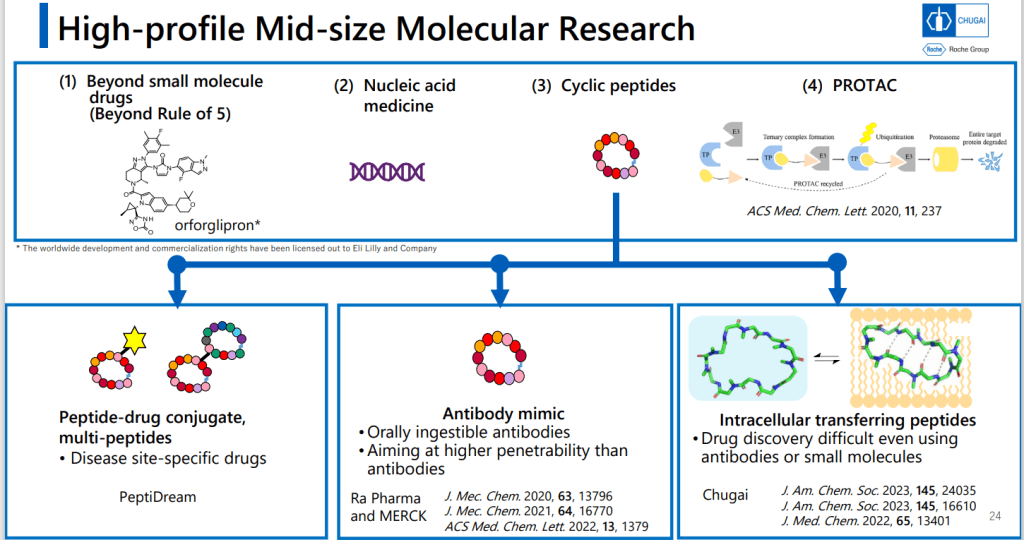
개념도를 보면 Antibody는 Membrane Receptor에 결합하는 방법이고 Small molecule의 특성을 가지면서 Antibody와 같은 선택성을 갖는 Mid-size molecule을 발견하는 것이 목표라는 것입니다.
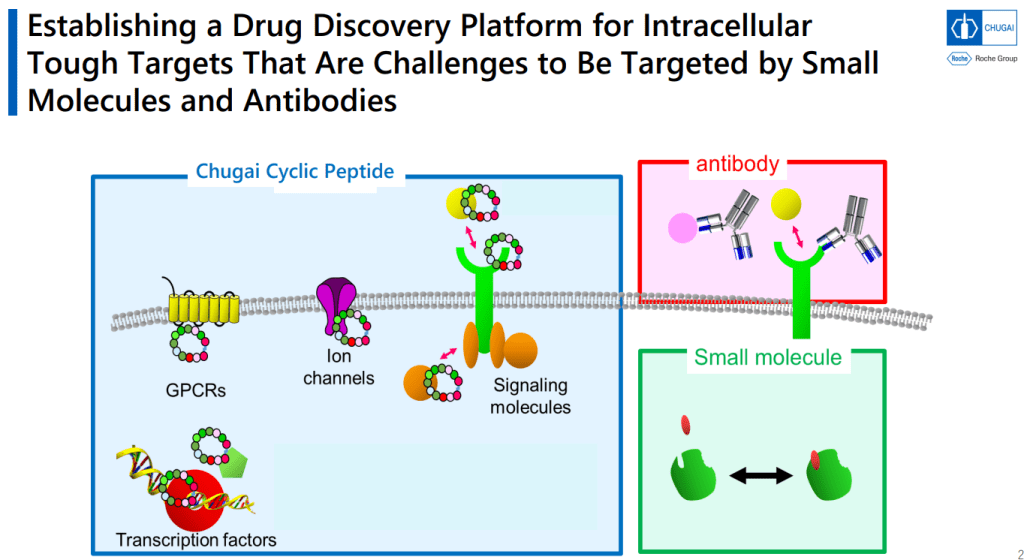
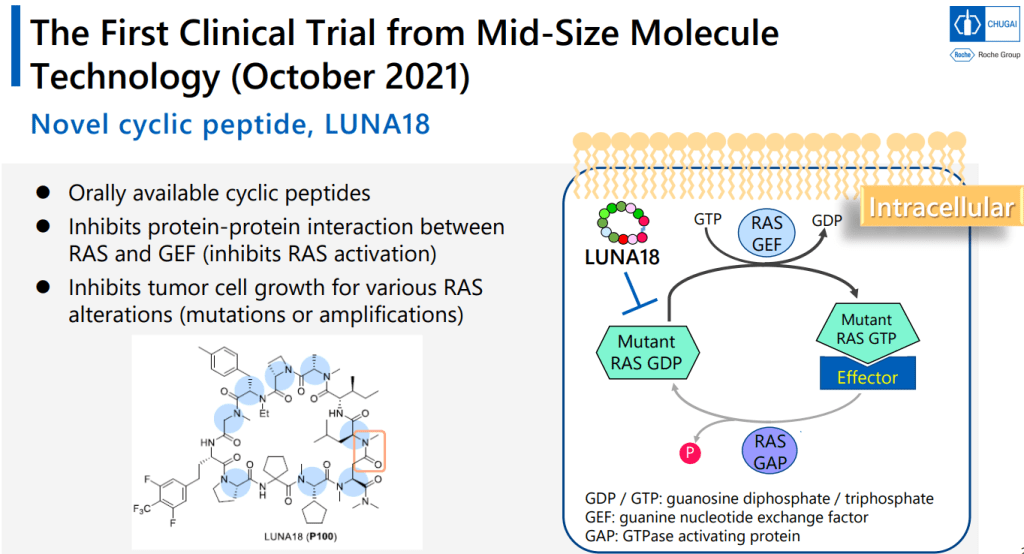
Mid-Size Molecule 중 처음으로 임상에 진입한 물질이 LUNA18입니다. pan-mutant RAS Inhibitor로 개발을 한 것입니다.
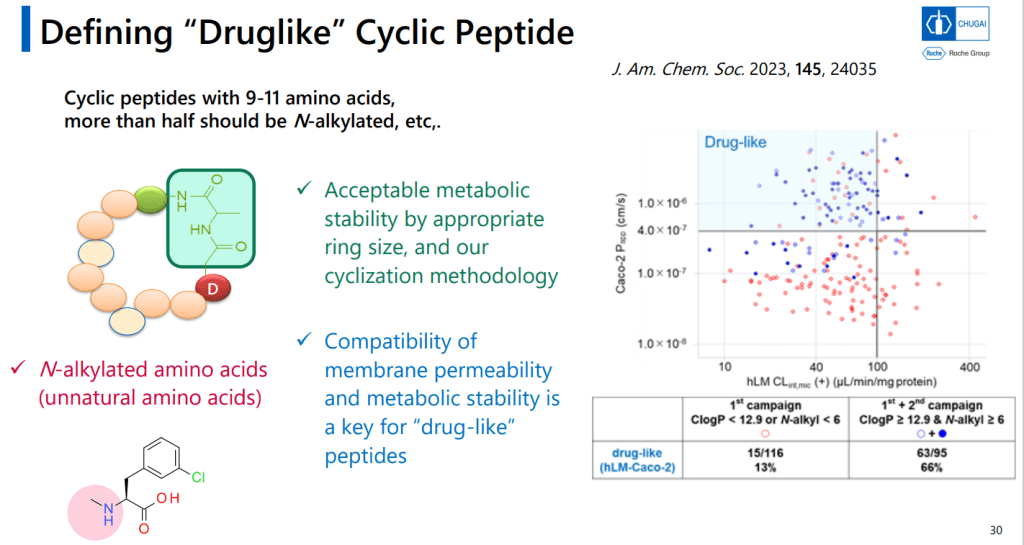
9-11-mer Macrocyclic Peptides가 Druglike한 Metabolical Stability를 위해서는 반 이상이 N-alkylated 되어야 한다는 것이 중요한 핵심 중 하나입니다.
PURESystem으로 Unnatural Macrocyclic Peptides를 만들 수 있는 mRNA Display Platform을 이룰 수 있었다고 발표했습니다.
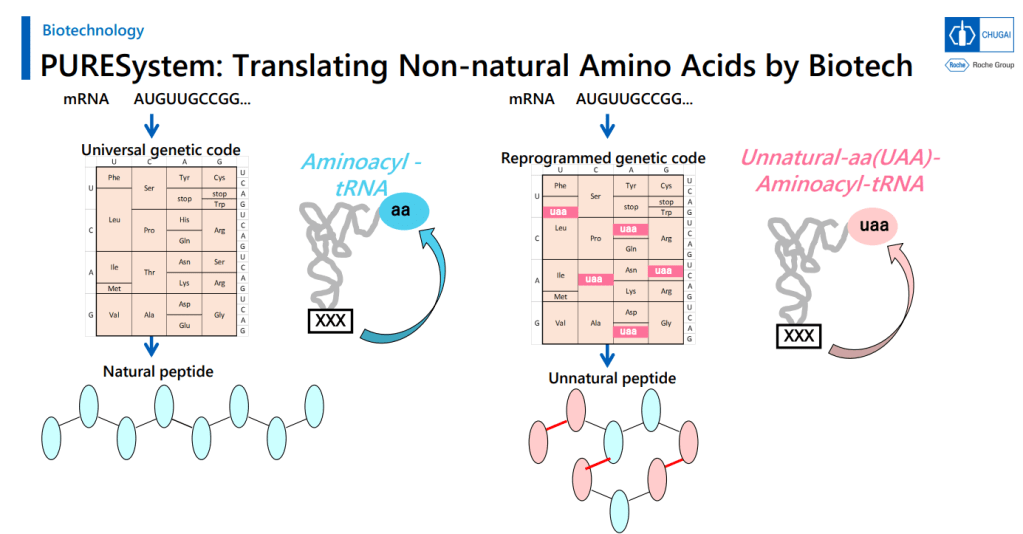
PURESystem에서 Cyclization Method가 Key Reaction이고 Cyclization 이후에 Desulfurization을 합니다.
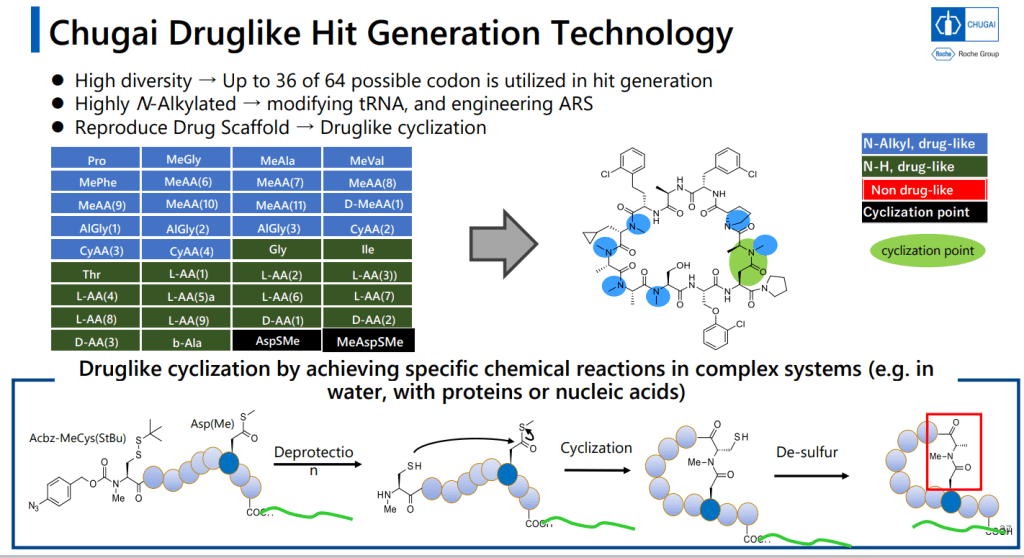
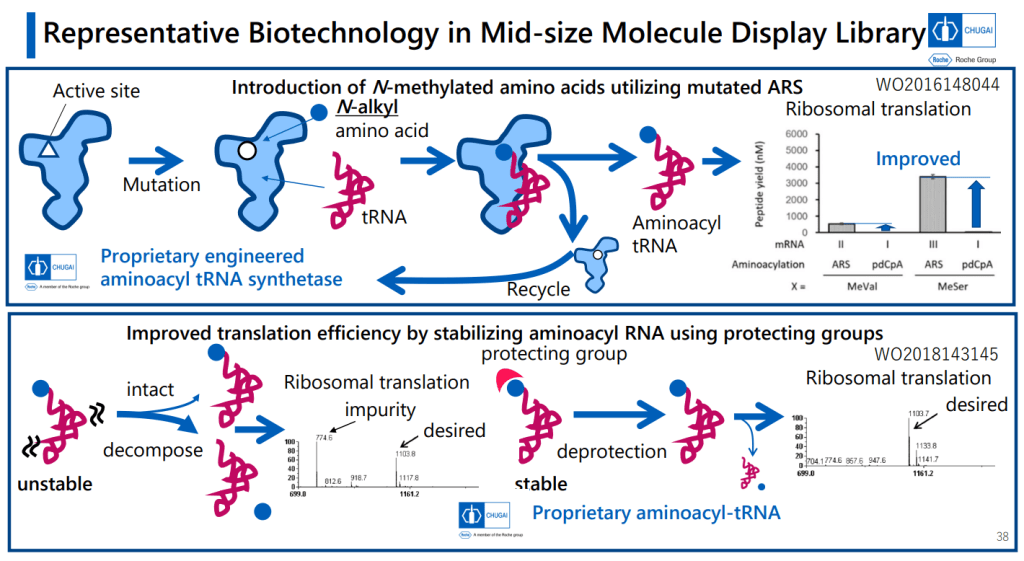
그리고 PeptiDream의 FIT (Flexizyme-mediated Flexible In Vitro Translation)이 N-alkyl amino acid를 받아들일 수 있도록 하기 위해 변형을 주었습니다.
BIOTECH (120) Peptidream: Flexizyme-FIT-RaPID Platform for Macrocyclic Peptides
Flexizyme을 mutation 시켰다는 것이 촛점입니다.
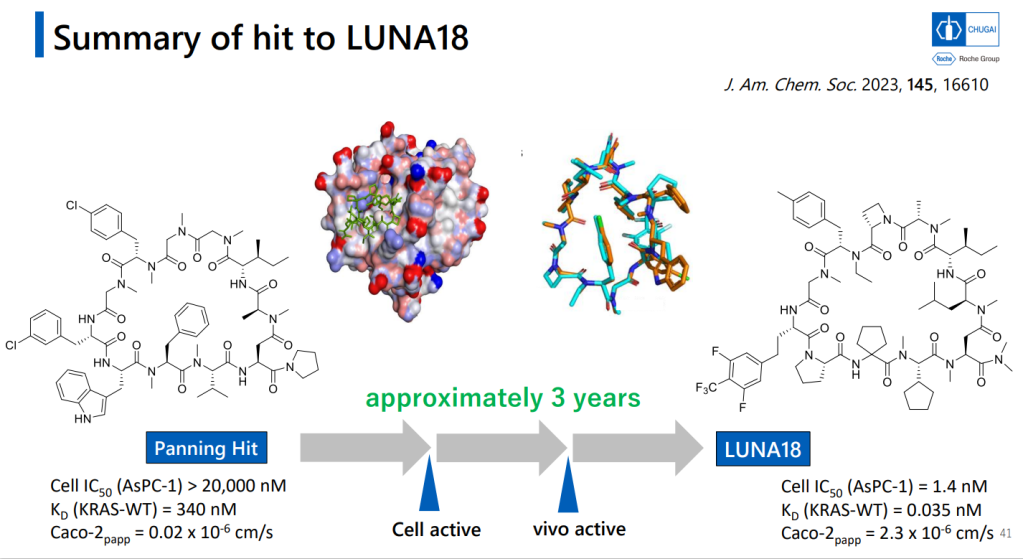
이렇게 다양한 Platform Technology를 이용했을 경우에 Hit Identification부터 Lead Optimization까지 약 3년의 시간이 걸렸습니다. 역시 Lead Optimization이 시간이 많이 걸립니다. Cell-based system에서 active molecule을 찾은 후 DMPK를 위한 Lead optimization을 나누고 있습니다.
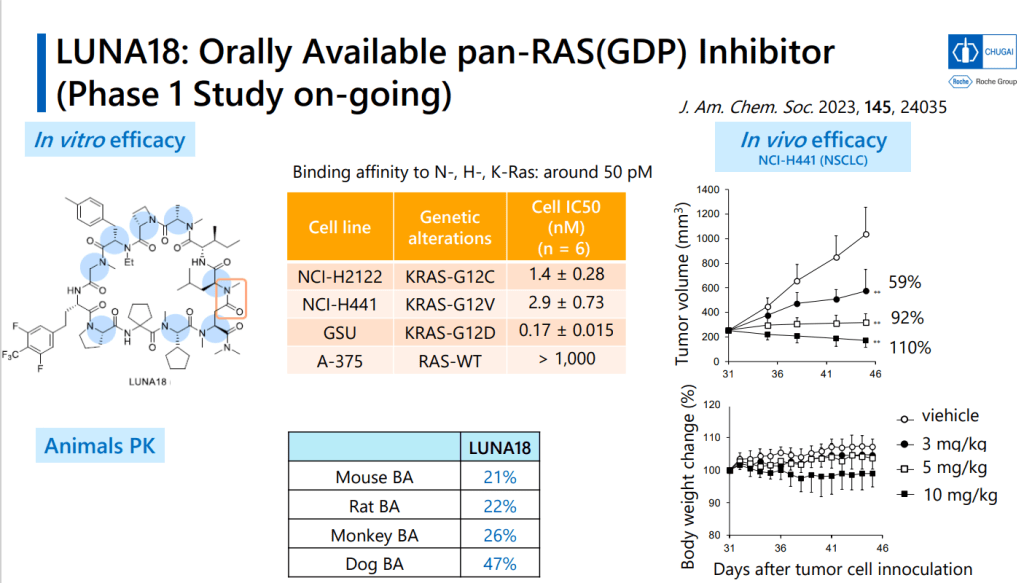
이러한 노력의 결과로 얻어진 LUNA18은 KRAS-G12C, KRAS-G12D, KRAS-G12V mutants에 모두 active하지만 KRAS-WT에서는 전혀 효과가 없기 때문에 정상세포에는 독성이 적고 Oncology KRAS mutants에만 듣는 Precision Oncology Medicine으로 될 가능성이 높은 약물입니다. In vivo efficacy도 dose에 따라 암세포가 줄어드는 것을 확인했고 체중은 대신 일정하게 유지가 되었습니다.
2023년 12월에 발표한 Chugai R&D Meeting Presentation은 아래에 링크합니다.
Novartis의 신약개발 Director이면서 Science에서 “In the Pipeline”이라는 블로그를 쓰는 Derek Lowe가 최근에 Chugai가 개발한 LUNA18 (pan-KRAS Inhibitor)에 대해 글을 쓴 것이 있습니다. Derek의 관점은 Chugai 연구팀이 N-alkyl amino acid를 이미 mRNA Display와 Solid-phase synthesizer에 도입을 해서 5번만의 In vitro selection만으로 180 nM active molecule을 찾았다는 점을 아주 높이 평가하고 있습니다. (Big Pharma Library Screening으로도 이런 것은 얻을 수 없다는 것을 강조하면서).
Chugai Pharmaceutical 연구팀이 Cell-Permeable Orally Active Macrocyclic Peptides를 위한 Druglike Platform을 한층 Upgrade한 것은 확실한 것 같습니다. 이 논문을 본 다른 연구팀들이 또한번 업그레이드 시키겠죠. 이 분야의 발전이 큰 기대가 됩니다.
Macrocyclic Peptide Drugs From the Ground Up – In the Pipeline by Derek Lowe 1/9/2024
I’ve been meaning to blog about this paper from a large team at Chugai, looking at ways to make rather large cyclic peptide structures that can also still be drugs. The whole “peptides as drugs” topic has been a perennial here on the blog, and by that I mean “going back to 2002“, with updates along the way. Here’s a recent review on the subject (and there are plenty more out there!)
The reasons it’s such a focus in drug discovery come from both ends of the topic. On the one hand, an awful lot of protein functions in the cell are mediated by, well, other proteins, or peptide pieces thereof. Protein-protein interactions (PPIs) are so ubiquitous, and the proteome that we have have is so tuned up for them, that a great many of our small-molecule drugs are actually fitting into binding sites that are normally part of some protein-binding event. (There are of course binding sites that are evolved for small molecules, such as with the amine GPCRs, and drug discovery efforts have naturally pounded away at those over the years, fear not).
But at the same time that there are a lot of protein-protein sites to exploit, actually getting down to expoiting them is difficult if you try to do it with an actual peptide, as opposed to some small molecule that ends up acting as a peptidomimetic instead. That’s because proteins of all sorts are constantly being recycled and remodeled in living cells. There are all varieties of saw blades spinning constantly in the biochemical environment, protease and peptidase enzymes that are ready to start slicing proteins up into smaller pieces. Our endogenous proteins are adapted to this, generally by being compartmentalized away from things that would chew them up and by not displaying easily-cleaved sequences to the enzymes they’re most likely to encounter. Instead, these protein-processing events are managed in a vast and intricate landscape, with a good example being the coagulation cascade.
Even getting to the stage where all these enzymes can take a whack at your peptide drug is not so easy, thanks to the way the digestive system is set up. We do not schlork up proteins as whole species when we eat them – instead, everything gets broken down thoroughly by digestive enzyme into individual amino acids, dipeptides, and tripeptides. Those are the species that are actually absorbed, and our innards are very good at ripping a huge variety of proteins into such sawdust. Which is what will happen to your drug candidate unless you take great care to avoid it.
There are more strategies than I can count for trying to fix these problems, and they have been refined and extended for decades now in drug discovery. N-methylation, reverse-chirality residues, beta-amino acids, “retro-inverso” chains, cyclic peptides of many kinds from simple rings to complex knots. . .those are some of the classics, and that’s just the start of the topic. The Chugai paper linked above is a contribution to this field, and a key step they’re taking is to start with the right sort of screening library – one that’s already most of the way to drug-likeness.
That means (they say) cyclic peptides, in roughly the 11-amino-acid size range, with more lipophilic side chains than usual, and a prominent amount of N-alkylation already built in. They’re taking their cues from cyclosporine, which is a notably effective compound with far better membrane penetration and pharmacokinetic behavior than one might have predicted. (Medicinal chemists have been mining the behaviors of such naturally-occurring macrocycles for a long time now!) The hope is that good hits from such a collection can be optimized without doing too much violence to the overall conformation of the ring and its physiochemical properties. Indeed, checking a library of 8-to-12-amino-acid membered ring cyclic peptide compounds showed that the 11-AA-membered ones had notably better stability to metabolic enzymes, and this is surely No Accident, evolutionarily. Similarly, it looks like you would want at least 6 alkylated residues (cyclosporine has 7) and a cLogP of at least 12.9 (cyclosporine’s is 14.4). Note: that is indeed quite greasy. And you’d like to have no more than 3 hydroxyl groups and a maximum of one ionizable group hanging off the structures as well (and probably none at all). Dosing a range of such compounds in mice confirmed that they were on the right track.
That’s a good amount of work already, but the group went on to work up an expression system to turn out large numbers of variants in this area for a screening library. That’s a challenge, because you’re asking the cellular protein synthesis machinery to do a number of things it normally doesn’t: you need it to handle plenty of N-alkylated amino acids, and what’s worse, you need to have some of these show up one after the other in the chains. You need to make macrocycles without relying on labile groups like disulfides or thioesters. And you need to strip out all the amino acids with ionizable side chains. That involves lot of engineering at the aminoacyl tRNA stage, but they managed to get an mRNA display system working with these modifications (using the PURE system, which is done outside of living cells and is thus more amenable to all the necessary changes).
mRNA display can give you a tremendous number of possible products, and indeed, the team ran an experiment that was capable of generating different peptides in the ten-to-the-tenth (tens of billions) range, and deploying this in a search for a KRAS ligand. That’s just the sort of audacious target this sort of technology should be applied to! Recent years have seen progress in targeting mutant forms of that cancer protein, but wild-type KRAS is a major challenge for anyone. After five rounds of enrichment, panning away all the less-potent candidates, they narrowed down on a particular cyclic peptide that had 180 nM activity in blocking the interaction of KRAS and its partner SOS1. You can screen the entire collections of big pharma companies and not find a compound like that (they have!)
The authors present an X-ray structure of the complex, which generally ends all arguments, and go on to show optimization of the compound into an even better drug candidate. As they’d hoped, this was achieved without any major alterations to the core structure, but rather relying on changing some side-chain properties. No polar (hydrogen-bonding) groups were used in that process – as they say, “it was not necessary to use polar functional groups to optimize the structure of a hit that does not rely on polar functional groups for binding to its target protein.”
These extra hydrophobic groups actually improved the properties of the new compound, which had sub-nanomolar activity for the KRAS-SOS1 interaction. It has good PK properties (47% oral bioavailability in dogs, for example), and is now in Phase I human trials. Which is pretty damned impressive (here’s another detailed look at the compound’s development). Chugai’s dedication to getting this macrocyclic-peptide-screening platform off the ground is impressive as well, and I very much look forward to seeing what else that can make out of it. And what the rest of the industry can make of the ideas behind it!
