 – 한미약품/Spectrum의 Poziotinib의 FDA 불승인 원인에 대한 생각")
안녕하세요 Boston 임박사입니다.
오랜만에 한국의 신약에 대한 얘기를 좀 해 보려고 합니다. 작년에 한국 제약 BIO 업계의 명암을 가른 약물 가운데 한미약품의 Poziotinib이라는 비소세포성폐암 (NSCLC) Ex20ins 치료제가 Accelerated Approval을 받았음에도 결국 US FDA 승인을 받지 못했습니다. 이에 대해 한국의 언론에서도 중요한 News로 다루기는 했는데요 하지만 승인을 받지 못하게 된 이유에 대해서는 어느 News에서도 아직 찾아보지를 못했습니다.
ODAC (미국 항암제자문위원회, Oncology Drug Advisory Committee)에서 9:4로 반대가 더 많았다는 것과 Enhertu 약물이 효과와 부작용 측면에서 좋았기 때문이라는 일반적인 기사는있었지만 진짜 이유에 대해서는 기사를 보지 못했습니다. 그래서 US FDA의 ODAC 내용과 US FDA의 불승인 결정을 번역하면서 짚어넘어가 보고자 합니다. 신약은 US FDA의 승인을 받을 수도 있고 받지 못할 수도 있습니다. 문제는 원인분석과 그 대응에 있다고 생각합니다.
먼저 한국 신문의 기사들 몇개를 올립니다.
스펙트럼, “美 FDA, 현 시점에서는 포지오티닙 승인 못 해” – 메디포뉴스 11/25/2022
미국 FDA로부터 “현 시점에서는 Poziotinib을 승인할 수 없다”는 내용의 CRL(Complete Response Letter)을 수령했다
미국 항암제자문위원회(Oncology Drug Advisory Committee)는 지난 9월 23일, US FDA의 시판허가 여부 결정에 앞서 Poziotinib이 환자에게 주는 현재의 혜택이 위험보다 크지 않다고 표결(9:4)한 바 있으며, 이번 US FDA의 결정은 당시 항암제자문위원회의 권고를 따른 것이다.
위의 메디포뉴스는 FDA의 입장을 온전히 전한 것 같지는 않은 것이 미국신문의 기사는 약간 뉴앙스가 다릅니다.
FDA Denies Approval of Poziotinib for Advanced/Metastatic NSCLC With HER2 Exon 20 Insertions – Targeted Oncology 11/28/2022
It is the position of the FDA that the NDA for poziotinib cannot be approved in its current state. 번역: Poziotinib의 NDA는 현재상태로는 승인할 수 없다는 것이 US FDA의 입장이다.
이 부분은 메디포뉴스와 같습니다. 문제는 다음이 중요한데 이 부분이 메디포뉴스에는 누락되었습니다.
Additional data are needed from a randomized controlled study for poziotinib to be approved for this indication, according to the FDA. 번역: FDA에 따르면 승인을 위해서는 Poziotinib의 무작위 배정시험을 통한 추가 Data가 필요하다.
즉, US FDA의 입장은 추가 임상 Data가 승인요건을 충족한다면 승인을 할 수 있다는 입장입니다. 이것은 지금 한미약품과 Spectrum이 제출한 임상 Data로는 승인을 할 수 없지만 US FDA가 요구한 추가 Data를 제출하면 다시 승인 가능성이 있다로 해석할 수 있는 것입니다.
그래서 저는 궁금해 졌습니다. 미국 항암제자문위원회 (ODAC)이 9:4 표결을 하게된 근거가 무엇인가?에 대해서 말이죠. 그래서 그 자료를 US FDA에서 Download 해서 가져왔습니다.
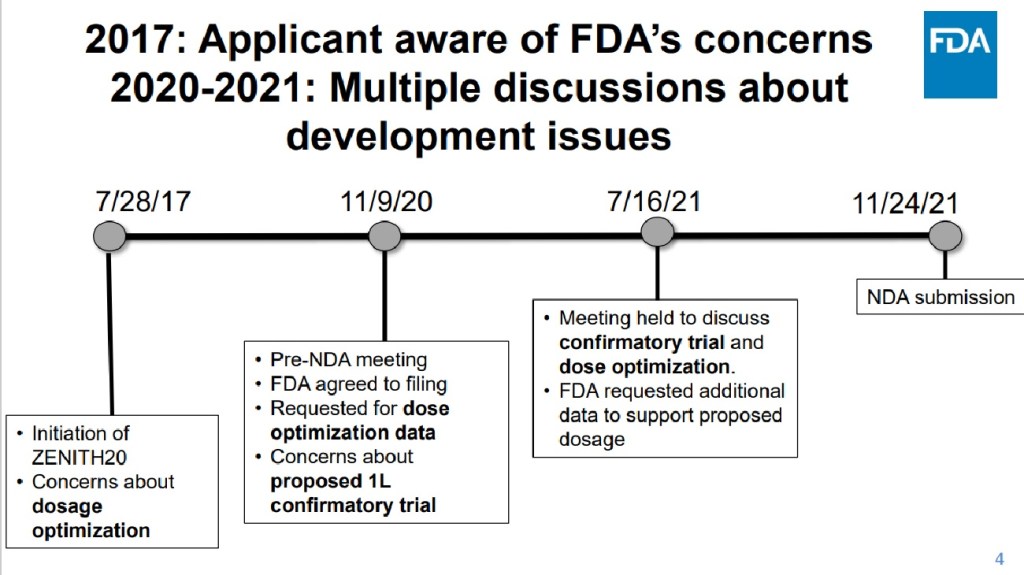
4번째 Slide에 보면 2017년부터 US FDA 미팅을 할 때마다 “용량 최적화 (Dosage Optimization)”에 대해서 계속 얘기를 했는데 지켜지지 않았다고 합니다.
2017년 7월 28일에는 용량최적화 우려 (Concern)이라는 표현이 나오고요.
2020년 11월 9일에는 용량최적화 Data를 요구했다 (Requested)와 함께 (회사가) 제안한 1L 확인시험에 대해 우려 (Concern)한다는 표현이 있습니다.
그리고 항암제자문위원회가 있기 2달전인 2021년 7월 16일에도 이런 내용이 있습니다.
Meeting 목적: 확정시험 (Confirmatory trial)과 용량최적화 (dose optimization)을 논의하기 위해서; 그리고
US FDA는 (회사가) 제안한 용량이 (적절한지를) 지원하는 추가 Data를 요구했다. 라고 나옵니다. 그러니까 이 항암제자문위원회는 2017년 이후 계속해서 현재 용량이 적절하지 않다는 우려와 추가 Data 요구를 해왔는데 회사는 이에 대해 제대로 응답을 못하고 4년을 끌어온 것입니다.
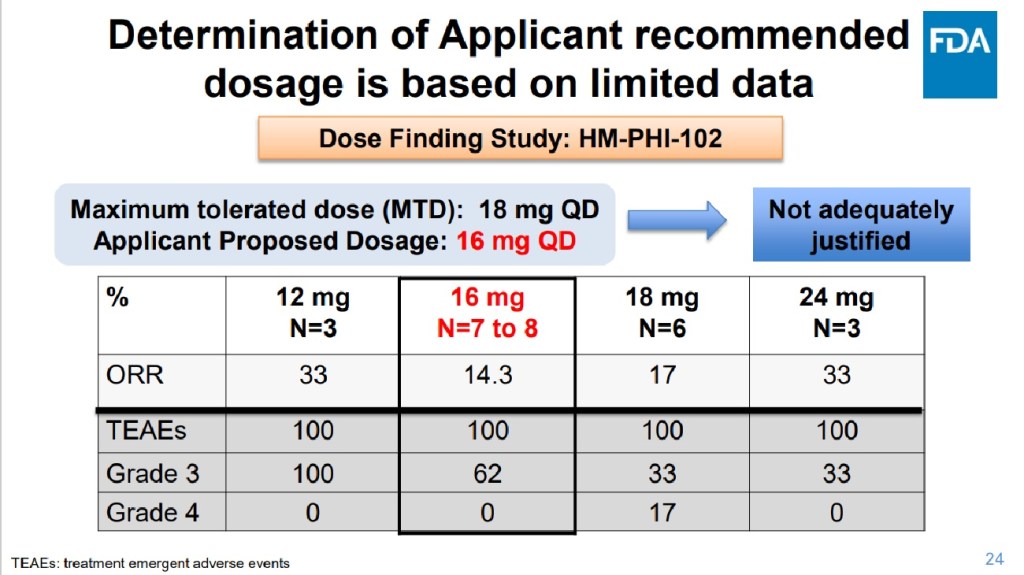
조금더 들어가서 그럼 US FDA와 항암제자문위원회는 왜 용량에 대해 우려를 했는지를 봐야겠죠. 다 얘기할 수 없는 관계로 Slide #24로 갑니다.
최대용량이 18mg QD였는데 회사가 제안안 용량은 그보다 2mg만 작은 16mg QD였고 이에 대해 “제대로 정당화되지 못했다 (Not adequately justified)”라고 나옵니다.
그리고 이에 대해 아래 붉은 색으로 16mg이 왜 정당화되지 못했는지를 보여주고 있죠. TEAE (치료응급부작용)이 100입니다. 그러니까 16mg을 복용하면 모든 환자에서 응급상황에 직면했다는 것입니다. 이 결과 때문에 US FDA는 16mg이 정당화되지 못한다고 하는 것입니다. 심지어 이보다 작은 12mg의 경우에도 100입니다. 그러니까 12mg보다 적어야 한다는 것이죠.
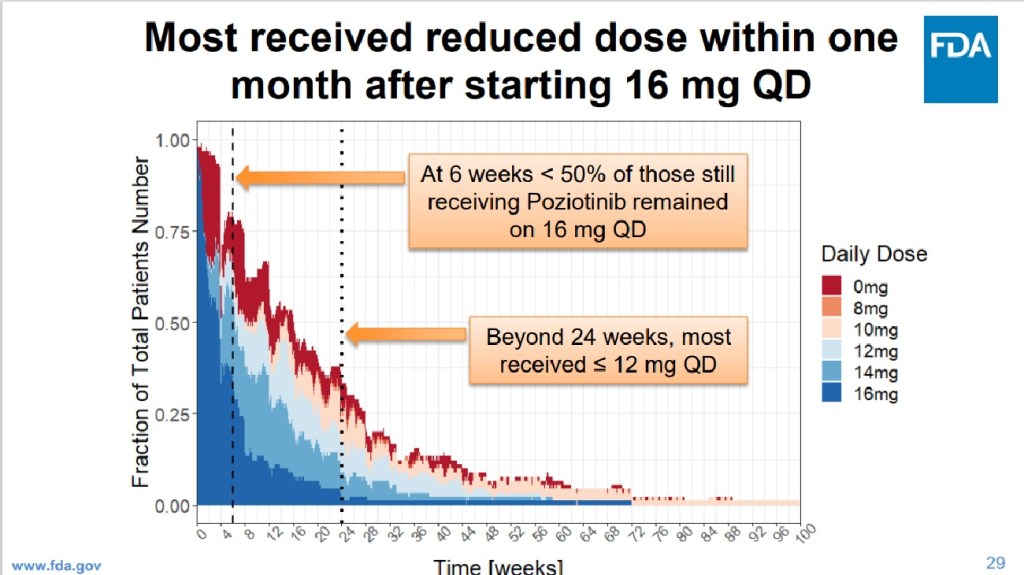
그리고 이것에 대해 Slide #27에서 정리를 해 줍니다. 환자들의 응급상황에 대해 설사 (diarrhea), 위염 (stomatitis), 용량을 줄여야 하는 응급상황이나 치료를 중단하는 상황이었다. – 따라서 적은 용량으로 안전성을 확보해야 한다.는 결론입니다.

이러한 US FDA의 우려는 임상시험에서도 그대로 나타났습니다. 24주가 지나면 대부분 12mg 이하로 용량을 줄였다는 것이죠. 그러니까 12mg 이하의 용량이어야 한다는 US FDA와 ODAC (항암제자문위원회)의 생각입니다.
참고로 Poziotinib과 같은 작용기전이면서 1년전에 승인받은 다케다제약의 Mobocertinib의 용량은 30mg 혹은 60mg QD입니다.
이미 기존에 다케다제약의 Mobocertinib이 있고 Enhertu가 효과와 안전성 문제에서 모두 우수하기 때문에 제가 보기에는 Poziotinib은 용량을 줄여야 하는데 용량을 줄이면 약효가 떨어질 우려가 있으므로 승인은 어려울 것 같습니다.
US FDA의 Back-Up Slides에 보면 지금의 용량으로도 약효가 가장 낮습니다. 데일리팜에서도 사실상 승인은 어렵다고 기사를 냈군요.
포지오티닙, 기술수출부터 美 진출 좌절까지 7년 스토리 – 데일리팜 11/28/2022
최근 Spectrum은 다른 회사에 인수가 되었습니다. 인수한 회사에서 Poziotinib에 투자를 해서 새로이 임상3상을 할 것 가능성은 매우 낮아 보입니다.
가정이지만 만약 Poziotinib이 먼저 승인받은 다케다의 Mobocertinib보다 앞서서 NDA를 했다면 얘기는 달라질 수 있습니다. 이미 승인된 약물이 있는 것과 전혀 없는 것은 US FDA의 심사에 영향을 주니까요.
여러모로 임상시험 속도가 늦었던 것도 원인이 될 것 같습니다. 아쉽네요.
