Productive Longevity에 대해 관심을 가지고 글을 쓰고 있는데 우리말로 번역하면 “생산적으로 오래삶”이라고 할 수 있을 것 같습니다. 즉, 노화라는 의미보다는 오래산다는 것에 방점을 찍는 용어라고 할 수 있죠. 노년학 (Gerontology)에서 노화에 대한 다양한 정의가 있는데 이것을 하나로 잘 정의해 주신 김수형님의 글이 있어서 아래에 나누고자 합니다.
나이 들어간다는 것은 우리에게 어떤 의미일까? 나이 들어간다는 것은 한편으로 늙어간다는 것이고, 신체적으로나 정신적으로나 성숙해져간다는 의미를 담고 있을 것이다. 이러한 나이 들어감, 한편으로 늙어간다는 것은 누구에게나 일어나는 필연적이고, 보편적인 현상이며, 어떻게 생각하면 가차 없이 발생하는 현상일 것이다. 이런 나이 들어가는 것과 늙어가는 것을 영어로는 ‘Aging’이라는 단어로 쓰인다. 한글로 번역하면 ‘고령화’, ‘노화’, ‘나이 먹음’을 의미한다. ‘노화’를 바라보는 시각은 비유적으로 컵 안에 반 정도 있는 물을 어떻게 바라보는지와 유사할 것이다.
긍정적인 시각으로 컵 안에 있는 물을 바라본다면 ‘아직도 물이 반이나 남았네!’라고 볼 것이고, 부정적인 시각으로 바라본다면, ‘물이 반 밖에 안 남았네!’라고 볼 것이다. ‘늙어감’을 바라보는 시각도 이와 비슷하지 않을까 묻고 싶다. 늙는 것을 긍정적인 시각으로 바라본다면, 나이 들어가는 것은 새로운 성장의 단계로 볼 수 있을 것이다. 여러 가지 단어가 떠올려진다. 낙관주의, 도전, 기회, 친밀감, 건강함, 목적, 열정 등 활동적이며 인생을 적극적으로 생활하는 액티브한 노년의 모습이 보인다. 하지만 그 반대로 생각하면 비관주의, 약함, 슬픔, 외로움, 두려움, 후회 등 부정적인 단어가 떠올려진다. 누구에게나 찾아오는 늙음을 여러분은 어떻게 바라보고 싶은가? 물론 개인적인 차가 있겠지만 단순히 나이로 생각하기 보다는 노화를 받아들이는 태도에 따라 노년의 모습도 달라지지 않을까 한다.
그럼 잘 늙어가는 모습은 어떤 것이 있을까? 우리 주변에서 찾을 수 있는 멋진 노년의 모습은 어떤 것이 있을까? 이에 대한 정답은 우리가 어떻게 노년의 모습을 정의하는지에 따라 다양하게 나타날 수 있다. 여기서는 8가지 늙어감의 모습을 제시하고 싶다.
첫째로 성공적 노화이다. 영어로 표현하면 ‘Successful aging‘이다. 성공적 노화는 ’질병과 장애가 없고, 인지적 기능과 신체적 기능을 유지하며, 적극적으로 인생참여를 지속하는 것‘으로 미국의 노년학자인 Rowe와 Kahn이 1987년에 발표한 논문에서 정의하고 있다. 신체적으로 건강하고, 인지와 정신적 기능을 유지하며, 사람과 사회 속에서의 관계를 잘 유지하는 것을 중시하고 있다.
둘째로는 활동적 노화이다. 보통 액티브 에이징(Active aging)이라는 영어 표현으로도 쓰인다. 활동적인 노년의 모습을 유지하기 위해서는 건강한 삶, 지속적인 사회참여, 경제적인 안정 등 3가지 조건이 충족될 필요가 있다.
세 번째로 긍정적으로 노년을 바라보는 모습이다. 영어로 ’Positive aging‘이라고 표현한다. 해외 유명인사들 중에 늙음을 긍정적으로 바라본 글귀가 여럿 있다.
미국의 작가이자 사회운동가였던 배티 프리던은 나이 들어가는 것은 ’잃어버린 젊음‘이 아니라, 새로운 기회와 힘의 단계라고 하였다. 미국의 유명 건축가였던 프랭크 로이드 라이트는 늙어가는 것을 “내가 더 오래 살수록, 더욱더 멋진 인생이 될 것”이라고 했고, 영화배우 아누크 에이미는 나이 들어가는 사람의 모습에서 진정한 아름다움을 찾을 수 있다고 했다.
네번째로 창의적인 노년의 모습이 있다. 창의적인 노화, 즉 Creative aging은 두 가지 측면에서 생각해 볼 수 있다. 하나는 사회참여와 기술습득에 초점을 맞춘 예술 프로그램에 적극적으로 참여하는 형태이고, 다른 하나는 지역사회 내에서 지속적으로 성장하고, 배우고, 공헌한다는 적극적인 사회 참여의 형태로 생각해 볼 수 있다.
다섯 번째로 젊게 살아가는 노년의 모습도 생각해 볼 수 있다. ’노노족‘이라는 말이 유행한 적이 있다. ’N0+老“, 즉 ‘노(NO)’와 ‘노(老)’를 합성해 만든 신조어이다. 건강 챙기기에 관심이 높고, 여행과 취미 활동에도 적극적이며, 외모에서도 실제 나이보다 훨씬 젊게 보이는 부류를 일컫는 말이다. 우리 주변에서도 젊은이 못지않은 왕성한 활동력을 보이고, 특히 젊은 층의 문화를 수용하려고 노력하는 젊은 노객을 본 적이 있을 것이다.
여섯 번째로 스마트(Smart)하게 노년을 살아가려는 모습이 나타나고 있다. 단순히 똑똑하게 나이 든다는 것이라고 해석하기 보다는 적극적인 배움을 통해 스스로 직접 노년을 디자인하는 모습을 스마트 에이징이라고 표현하고자 한다.
일곱 번째로, 현대 사회에 기술적 진보에 따라 디지털을 활용하는 노년의 모습이 나타나고 있다. 흔히, 디지털 에이징(Digital aging)이라고 표현한다. 정보통신기술(ICT)을 활용해 건강하고 활동적으로 나이 든다는 것을 의미한다. 디지털 기기에 친숙하지 않은 노년층을 위해 스마트폰을 사용하는 법을 알려주는 교육이나 노년층 대상 컴퓨터 강좌가 최근 성황리에 운영되는 이유이기도 하다.
마지막으로 생산적 노화의 모습이다. 영어로 표현하면 Productive aging이다. 긍정적인 노년의 모습을 통해 개인이 자신의 삶과 지역사회에서 중요한 공헌을 할 수 있다는 부분을 강조한 접근방식으로 해석된다. 일의 맥락에서 생산적 노화를 살펴보면, 근로자가 나이가 들어서도 그 기능을 유지하고 발휘될 수 있도록 모든 사람에게 안전하고 건강한 근로 환경을 제공하는 것을 포함한다는 것을 의미한다.
지금까지 여덟 가지로 해석된 다양한 늙어감의 모습을 살펴보았다. 각각의 노화의 모습이 의미를 담고 있는지 설명한 내용이었다. 어떤 면으론 늙어감의 모습이 진화된다고 해석할 수 있다.
우리 주변에 있는 고령자를 볼 때 어떤 노년의 모습으로 대입될 지 찾아보는 것도 흥미로운 일일 것이다. 아마 위에 포함된 노년의 모습을 많이 발견할 때….점차 노년이 행복한 사회로 되지 않을까?
Once considered an undruggable target, KRAS now has two FDA-approved therapies vying for a blockbuster cancer market.
Mirati Therapeutics’ Krazati, also known as adagrasib, will take on Amgen’s first-to-market Lumakras thanks to an FDA accelerated approval in previously treated KRAS G12C-mutated non-small cell lung cancer (NSCLC).
Krazati marks Mirati’s first commercial product, and the biotech will focus on efficacy in its marketing pitch, CEO David Meek told Fierce Pharma in an interview ahead of the approval. J.P. Morgan analyst Eric Joseph, Ph.D., has in late November put Krazati’s risk-weighted peak sales estimate across multiple indications at $1.7 billion but then lowered the number to $1.3 billion a few days ago, as blockbuster hopes for the drug have dwindled since a Keytruda combination readout.
Mirati is charging Krazati at a list price of $19,750 for a 30-day supply, a company spokesperson told Fierce Pharma. Krazati is given in 600-mg capsules twice daily. By comparison, Lumakras costs slightly less with a list price of $17,900 per month
In the phase 2 registrational portion of the KRYSTAL-1 trial, Krazati shrank tumors in 43% of patients when used in patients with previously treated NSCLC bearing KRAS G12C mutations. By comparison, Lumakras, given 960 mg once daily, showed a 36% tumor response rate in its own phase 2 trial. And the number dropped to 28% in a larger phase 3 trial.
Similar to Lumakras’ situation, the FDA has required Mirati to run a postmarketing study to test a lower 400-mg twice daily regimen of Krazati. The FDA’s oncology department has recently put an emphasis on dose optimization under Project Optimus. The agency has criticized drugmakers for pushing for the highest tolerable dose in early clinical testing without carefully examining the benefit-risk balance.
About 25% to 50% of NSCLC cases develop brain metastases during the course of the disease. In another key component of Mirati’s commercial campaign, Krazati has shown a brain tumor response rate of 33% versus 25% for Lumakras in patients with baseline brain metastases in its own study. A label with that brain metastases information would be a “nice-to-have” that can differentiate Krazati from the competition, but the data are not included, J.P. Morgan’s Joseph pointed out in a Tuesday note.
In a pooled analysis of the phase 1/2 KRYSTAL-1 trial, patients on Krazati lived a median 14.1 months. By comparison, Lumakras takers survived a median 12.5 months in its phase 2 trial and 10.6 months in the phase 3 study. Krazati’s own phase 3 confirmatory trial, KRYSTAL-12, which compares Krazati with the chemotherapy docetaxel, is underway.
Cross-trial comparisons come with intrinsic problems such as patient characteristics differences. But Meek stressed that Krazati’s response data are best-in-class so far.
In what Meek calls a “halo” effect, Mirati also hopes doctors will notice the company’s recent early results for Krazati’s combination with Merck’s PD-1 inhibitor Keytruda. The early-stage KRYSTAL-7 trial showed what Barclays analysts called “good safety but modest activity” in newly diagnosed NSCLC. In contrast, Merck’s Keytruda combo reported lackluster efficacy data while raising a serious liver safety concern.
“I think we’re the KRAS leader,” Meek said in the interview. “We will set the direction where this KRAS agent goes.”
But while Mirati’s KRAS-Keytruda combo data looked better than Amgen’s, Krazati’s commercial potential is under question as well. The company’s stock price has been about halved after the KRYSTAL-7 combo study, reflecting investors’ concern that the companies’ planned phase 3 trials might not succeed.
Although Krazati is Mirati’s first commercial launch, it’s not the first for the Mirati people. The company has built a sales force with average 19 years of experience in oncology, and the staffers were hired based on their experience in lung cancer, Meek noted.
Mirati has talked to nearly all of the top payer plans and received positive feedback, Chief Commercial Officer Ben Hickey told investors during the company’s third-quarter earnings call last month. Mirati expects to have broad coverage within the first few months of launch, he said.
Krazati’s launch comes as Lumakras experienced a sequential sales decline in the third quarter to $75 million. Amgen attributed the slowdown to a price adjustment as part of reimbursement deal in Germany.
Next up, Mirati plans to launch two phase 3 trials of Krazati—at the 400-mg twice daily dose—in combination with Keytruda in front-line NSCLC this year. The company is discussing a potential accelerated approval pathway for Krazati in third-line colorectal cancer and will have more to share in early 2023, Meek said. The confirmatory KRYSTAL-10 trial for a cocktail of Krazati and Eli Lilly’s Erbitux in second-line colorectal cancer is also expected to read out later next year. In addition, the company has a candidate for KRAS G12D mutations that’s slated to enter clinical testing next year.
With all the pipeline advancement, Mirati has recently reportedly attracted buyout interest from Big Pharma. Mirati has tapped an adviser, and larger pharmas are considering the “merits of a transaction,” Bloomberg reported late November.
“We’re real busy,” Meek said in his interview with Fierce Pharma. “We’re really focused on executing our clinical plans and our launch plans.”
However, any potential acquirer might have pulled out by now, BMO Research analyst Evan David Seigerman said in an investor note after the KRYSTAL-7 combo readout. The phase 3 combo trials Mirati plans to run will take a long time to read out, and existing early-stage data don’t bode well, he said.
Bristol-Myers Squibb (BMY.N), opens new tab on Sunday said it will acquire cancer drugmaker Mirati Therapeutics (MRTX.O), opens new tab for up to $5.8 billion, diversifying its oncology business and adding drugs it hopes can help offset expected lost revenue from patent expirations later this decade.
Bristol will pick up Mirati’s portfolio drugs that target the genetic drivers of specific cancers including its lung cancer drug, Krazati, which was approved in December.
A second compound – MRTX1719 – which could be used in some types of lung cancer was also attractive to the company, Bristol executives said in an interview.
“We think this really helps strategically complement our oncology portfolio but also, from a financial standpoint, it helps out commercially in the back half of the decade,” said Adam Lenkowsky, Bristol’s Chief Commercialization Officer.
The company said that it will buy Mirati for $58 per share in cash, or around $4.8 billion. Mirati has around $1.1 billion in cash on hand, so “we’re paying essentially $3.7 billion enterprise value…we think with that we’ve gotten a very attractive deal,” Lenkowsky said.
Mirati stockholders will also receive one non-tradeable contingent value right for each Mirati share held, potentially worth $12.00 per share in cash, representing an additional $1 billion of value opportunity, the company said
Bristol will finance the transaction with a combination of cash and debt, the company said in a statement.
The U.S. Food and Drug Administration in December approved the drug to treat adults with advanced lung cancer.
“With multiple targeted oncology assets including Krazati, Mirati is another important step forward in our efforts to grow our diversified oncology portfolio and further strengthen Bristol Myers Squibb’s pipeline for the latter half of the decade and beyond,” said Chris Boerner, Bristol’s incoming CEO and current chief operating officer, in a statement.
The New York-based company has been pressured by declining demand for two of its top drugs, the blood cancer treatment Revlimid and blood thinner Eliquis, which face generic competition.
Bristol is buying Mirati at a time when the shares are considerably cheaper than they were. Mirati’s shares touched a 52-week high of $101.3 apiece on Nov. 28 and are now trading at $60.2.
The transaction is expected to be dilutive to Bristol’s non-GAAP earnings per share by approximately 35 cents per share in the first 12 months after the transaction closes, the statement added.
In April, Bristol said CEO Giovanni Caforio would step down in November and be succeeded by Boerner.
Last year, Bristol acquired drug developer Turning Point Therapeutics for $4.1 billion in cash to help bolster its arsenal of cancer drugs.
Bristol Myers Squibb looks on track to overtake Amgen as the KRAS leader in lung cancer after following up its rival’s FDA setback with a positive confirmatory trial readout.
BMS’ Krazati significantly reduced the risk of tumor progression or death compared with chemotherapy in patients with pretreated KRAS G12C-mutated non-small cell lung cancer (NSCLC), the company said Thursday. The KRAS inhibitor came to the New Jersey pharma as part of its recent acquisition of Mirati Therapeutics.
The assessment was made by a blinded central review committee of the pivotal phase 3 KRYSTAL-12 study, which serves as the confirmatory trial for Krazati’s accelerated approval as a second-line therapy. BMS said it’s finishing a full evaluation of the data and will share results with regulators.
Besides declaring that the trial met its primary endpoint of progression-free survival, the independent data reviewers noted that Krazati was better than chemo at shrinking tumors, which was one of the trial’s secondary endpoints. The improvements on both endpoints were statistically significant and clinically meaningful, according to BMS.
The trial remains ongoing to evaluate whether Krazati can extend patients’ lives. BMS didn’t specify which direction Krazati’s survival outcomes are trending right now. Progression-free survival has typically been an approval-worthy endpoint in second-line NSCLC, unless there’s a negative trend in overall survival.
As for Amgen, the California drugmaker recently applied for full approval of its first-to-market KRAS inhibitor, Lumakras, based on progression-free survival data from the phase 3 CodeBreaK 200 trial. The study would have served its purpose had it been done properly. But the FDA figured its results couldn’t be reliably interpreted, and a group of external advisers agreed.
The agency and its advisory committee experts voiced concerns about disproportionate patient dropout rates between the two trial arms in Lumakras’ CodeBreaK 200 study as well as a bias for investigators to be more likely to call tumor progression early for patients on chemo so that they could cross over to receive Lumakras.
Both problems were chalked up to the enthusiasm around Lumakras as the first FDA-approved KRAS inhibitor.
By comparison, Krazati’s KRYSTAL-12 requires confirmation from a blinded central review to determine tumor progression before crossover.
Despite the compromised trial results, the FDA has let Lumakras stay on the market while Amgen runs another confirmatory trial to be completed no later than February 2028. The recently launched phase 3 CodeBreaK 202 trial is comparing Lumakras against Merck’s Keytruda in their respective combinations with chemotherapy for patients with newly diagnosed, advanced, PD-L1-negative, KRAS G12C-positive nonsquamous NSCLC.
Lumakras’ setback gives Krazati an opportunity to catch up. Before the BMS buyout, Krazati in the third quarter posted $16.4 million in sales, coming in below analysts’ expectations. Lumakras generated $52 million sales during the same period.
BMS’ Krazati also appears to hold more potential in the first-line setting. While Amgen was forced to combine Lumakras with chemo alone, a better liver toxicity profile has allowed BMS to pair Krazati with drugs in the PD-1 inhibitor class. A phase 3 trial is testing the Krazati-Keytruda combo in first-line KRAS G12C-mutated PD-L1-high NSCLC. And the company expects phase 2 results this year to guide its development path in PD-L1-low disease.
Both BMS and Amgen are also gunning for approvals in colorectal cancer, which is a smaller market than NSCLC. Amgen recently reported that Lumakras, at its currently approved 960-mg dose and used in combination with Vectibix, extended the median progression-free survival to 5.6 months versus 2.2 months for standard treatments in patients with chemo-refractory KRAS G12C colorectal cancer. The result came from the phase 3 CodeBreaK 300 trial.
A regulatory submission based on the study was planned in the first half of 2024, Amgen said during its fourth-quarter report. The company recently also launched a phase 3 trial for Lumakras in combination with Vectibix and chemo in first-line colorectal cancer.
For its part, BMS has the phase 3 KRYSTAL-10 study for Krazati and Eli Lilly’s Erbitux in second-line colorectal cancer, with a readout expected this year.
Themis Bioscience, a Vienna, Austria-based biotechnology start-up developing vaccines to prevent infectious diseases, has completed a €5m Series A financing round.
The round was co-led by Ventech and Crédit Agricole Private Equity.
The company intends to use the funds to advance the pre-‐clinical and clinical development activities for its lead product candidates, a Dengue and a Chikungunya Fever vaccine, which are both based on a novel vaccine vector technology (Themaxyn™) that was initially developed at the Institut Pasteur in Paris.
Founded in 2009 and led by CEO Erich Tauber, Themis Bioscience received seed financing by the academic business incubator INiTS, the austria wirtschaftsservice (aws) and The Austrian Research Promotion Agency (FFG).
VIENNA, Austria I March 02, 2015 I Themis Bioscience (‘Themis’), a biotechnology company developing innovative prophylactic vaccines for emerging tropical infections, and the Institut Pasteur, an international biomedical research center based in Paris (France) today announced the publication of the phase I study results for a recombinant measles-virus-based chikungunya vaccine (MV-CHIK) in The Lancet Infectious Diseases. The study was performed in collaboration with the Department of Clinical Pharmacology at the Medical University of Vienna and the Viral Diseases Branch of the Walter Reed Army Institute of Research (WRAIR) in the USA.
The peer-reviewed article is entitled “Immunogenicity, safety, and tolerability of a recombinant measles-virus-based chikungunya vaccine: a randomised, double-blind, placebo-controlled, active-comparator, first-in-man trial“.
Chikungunya fever is a mosquito-borne viral disease causing symptoms including fever, headache, joint and muscle pain and bleeding of the nose and gums. Importantly, a large number of infected patients suffer from chronic sequela months and years after the acute infection. The chikungunya virus originated in Asia and western and central Africa and rising levels of travel and global warming led to increasing incidences of the disease in temperate zones, thus becoming a global health threat. Since late 2013, more than one million cases have been reported in the Americas and the Caribbean alone, resulting in a significant public health and economic burden.
Themis’ recombinant measles-chikungunya vaccine phase I study was conducted between November 2013 and June 2014 with a total of 42 healthy male and female individuals from age 18-45 being randomised into 4 cohorts for this dose escalation study. Subjects were administered one injection with either a low, medium or high dose of the chikungunya vaccine or the active comparator Priorix (standard measles vaccine). The study investigated the immunogenicity, safety and tolerability of the vaccine. In addition, randomized participants received a booster injection on either day 28 or day 90 after the first vaccination.
The candidate vaccine raised concentrations of neutralising antibodies to chikungunya in all dose cohorts after one immunisation, with seroconversion* rates of participants producing anti-chikungunya antibodies of 44% in the low, 92% in the medium, and 90% in the high-dose group. The immunogenicity of the candidate vaccine was not affected by pre-existing anti-measles immunity. The second vaccination resulted in a 100% seroconversion for all participants in the candidate vaccine groups. The candidate vaccine had an overall good safety profile, and while the rate of adverse events increased with vaccine dose and volume, no vaccination-related serious adverse events were recorded.
Dr. Frederic Tangy, head of the Viral Genomics and Vaccination Unit at the Institut Pasteur (Institut Pasteur, CNRS UMR-3569), who developed this vaccine technology, explained: “The measles vaccine has already proven its high efficacy and safety on more than a billion vaccinated individuals during the last 30-40 years. Therefore, this platform offers an excellent safety profile and the clear advantage of a validated and easy manufacturing process. The present result demonstrates that a measles vector can be used in the presence of pre-existing immunity to measles, likely because it is a replicating vector. This gives another great advantage to this vaccine strategy.”
“Recent outbreaks have raised awareness of chikungunya virus worldwide and whilst further work is needed to show safety, tolerability, and ability of the vaccine to protect against live chikungunya virus, our trial data suggest that this novel vaccine is an excellent candidate to help address this urgent medical need”, explains Dr. Erich Tauber, CEO of Themis. “With these promising results we are advancing the chikungunya vaccine programme and aim to move rapidly into phase II studies.”
* seroconversion rate: percentage of participants/patients that produce antibodies
About Themis:
Themis Bioscience GmbH develops prophylactic vaccines with a focus on emerging tropical infectious diseases and has initial vaccine candidates currently in development for chikungunya and dengue fever. The company has exclusive, worldwide licenses for chikungunya- and dengue vaccines, based on the innovative and fully patent-protected measles virus vaccine vector platform from the Institut Pasteur in Paris. This platform underpins Themis’ growing pipeline of vaccines. Themis and Institut Pasteur are actively collaborating on additional targets. Themis was founded by experienced vaccine experts in September 2009 and is based in Vienna. For more information, visit the website: http://www.themisbio.com
Themis Bioscience (‘Themis’), a biotechnology company developing innovative prophylactic vaccines for emerging tropical infections, announced today the first closing of EUR 7 Million in a Series B financing of up to EUR 10 Million, led by new investor Wellington Partners. Existing investors Ventech and Omnes Capital (formerly Crédit Agricole Private Equity) also participated in the round. Dr Regina Hodits, General Partner at Wellington Partners will join Themis’ Board.
With their Chikungunya fever vaccine candidate demonstrating good immunogenicity, safety and tolerability in a Phase 1 clinical trial (Results published in The Lancet Infectious Diseases, March 2015), Themis plans to progress this lead product candidate into clinical phase II trials. In parallel the company will advance its other promising development pipeline in collaboration with the Institut Pasteur in Paris, originators of the measles vector platform licensed to Themis.
Themis also announced the new structure of its Board with Dr Gerd Zettlmeissl being named Chairman of the Board. Dr Zettlmeissl spent more than 20 years in executive positions in the international pharmaceutical and vaccine biotech industry. From 2005 until May 2011 he served as CEO of Intercell AG. Experienced biotech and vaccine industry expert Dr Jean-Paul Prieels will join as a new member the expanded board. He served as Senior Vice President of R&D at GlaxoSmithKline Biologicals until January 2011, led GSK’s global vaccine R&D development activities and was Head of Research for GSK Vaccines.
Dr Erich Tauber, CEO of Themis stated: “With the new funds, we are planning to move our Chikungunya vaccine candidate quickly into a Phase 2 clinical trial and also achieve important milestones for the other vaccine candidates in our preclinical development pipeline. We are very pleased to have Wellington Partners leading this financing round and I would like to welcome Dr Regina Hodits and Dr Jean-Paul Prieels to the Board. I am sure that Themis will profit from their scientific and industrial expertise in supporting our goals to establish new partnerships within the pharmaceutical industry and to drive our product pipeline towards commercialisation.”
Dr Regina Hodits, General Partner at Wellington Partners, commented: “With global warming and increased travel activities, tropical diseases like Chikungunya, Dengue fever, and other viral diseases are becoming a serious threat to global health. Based on a proven measles vaccine platform, Themis’ portfolio of vaccine candidates represent an attractive investment opportunity for Wellington, and they have the potential to address urgent unmet medical needs.”
In 2011, Themis raised EUR 5 Million in a series A financing following a seed financing round from Austria Wirtschaftsservice (AWS) in 2009, and other substantial financial contributions from Austrian national funding agencies like FFG and Inits.
About a year after a €10 million series B, Austrian vaccine company Themis has secured a series C in the same amount led by new investor Global Health Investment Fund (GHIF).
The money will again be used to advance a chikungunya vaccine, which is being tested in three phase 2 trials in central Europe, Puerto Rico and U.S. mainland, a Zika candidate that entered human testing last April, as well as other preclinical assets against RSV and norovirus. These vaccines are based on Themis’ proprietary Themaxyn platform developed at Institut Pasteur, which uses a measles vaccine as a vector to carry antigen-encoding genes.
Themis recently reported positive interim results from the European trial on its chikungunya vaccine, the most advanced program in its pipeline. The vaccine induced neutralizing antibodies in all treatment groups 56 days after first immunization, and the seroconversion rate reached 95% after two doses.
The European trial will have final readouts midyear, but CEO Erich Tauber, Ph.D., told FierceVaccines that the U.S. and Puerto Rico trials were delayed by hurricanes Harvey and Maria. The program also received £3 million worth of funding from the U.K.’s National Institute for Biological Standards and Control to develop a monkey challenge model and to conduct a small phase 1 in the country.
New investor GHIF led the round because it sees “tremendous potential in Themis’ technology platform” and is “impressed with Themis’ ability to navigate complex clinical, regulatory and manufacturing issues,” commented GHIF partner Glenn Rockman, who has joined Themis’ board.
While at J.P. Morgan’s investment banking division, Rockman worked with the Bill & Melinda Gates Foundation to build GHIF. The fund is focused on late-stage projects in drugs, vaccines and diagnostics for diseases that burden low-income populations. It has supported projects in malaria, tuberculosis, HIV/AIDS, cholera and preventable causes of maternal and infant mortality.
No vaccine is available for either chikungunya or Zika, both mosquito-borne viruses. In chikungunya, Themis is notably vying against PaxVax, which in-licensed its candidate from the NIH, and India’s Bharat Biotech and secretive Moderna are working on their phase 1 programs. More candidates are competing in Zika, including one from Inovio, a U.S. Army-developed shot Sanofi recently walked away from, and one from a Valneva-Emergent BioSolutions partnership, among others.
The Coalition for Epidemic Preparedness Innovations (CEPI), the high-profile public-private vaccine initiative launched in 2017, has signed its first company agreement, granting Themis an investment of up to $37.5 million to develop new vaccines against Lassa fever and MERS.
The grant spans a five-year period and will support Themis through phase 2 testing, providing safety and immunological data plus the manufacturing of investigational supplies for efficacy trials or emergency deployment during outbreaks.
Discoveries made by Institut Pasteur and the Paul Ehrlich Institut are set to become the basis for Themis’ Lassa and MERS candidates, respectively. Those two research institutions have identified antigens for inclusion in vaccine compositions and have demonstrated proof of concept in animal studies, Themis CEO Erich Tauber told FierceVaccines.
Themis will apply its measles vector platform—which it exclusively licensed from Institut Pasteur—to design the vaccines. The platform has been used in the company’s lead program for Chikungunya, which is in phase 2 trials in 600 patients in the U.S. and Europe. Its Zika candidate also uses the platform and has entered human testing, while other assets against norovirus, RSV and CMV are in preclinical stages.
“The fact that Themis has developed a versatile technology platform for the discovery, development and production of vaccine approaches is very attractive,” CEPI spokeswoman Rachel Grant told FierceVaccines. “This means as well as focusing on MERS and Lassa we hope this technology will have value beyond those specific diseases. ”
No additional financial details were disclosed, but given CEPI’s founding principle of equitable access, Grant said the agreement contains provisions that support providing vaccines at affordable costs to people in need.
CEPI focuses on epidemic vaccine development, especially where there’s unfavorable market incentives but potentially big public health benefits. The idea is to fund promising vaccine candidates so that they’re available immediately when an outbreak begins.
Officially launched in 2017 by governments and nonprofits such as the Bill & Melinda Gates Foundation and Wellcome Trust, the group has also attracted major vaccine makers including GlaxoSmithKline, Merck, Johnson & Johnson, Pfizer, Sanofi and Takeda. It has so far collected $630 million of its $1 billion target funding. The European Commission has also promised a contribution of €250 million that will support relevant projects through its own mechanisms.
To start, CEPI selected Lassa, MERS and Nipah as initial diseases to target, none of which have approved vaccines. The coalition actually aims to develop two promising vaccine candidates against each of these diseases, and Grant said CEPI is going through intensive technical and legal due diligence with a number of companies to finalize additional agreements over the coming months.
For a second project that will identify platforms for rapid vaccine development against unknown pathogens, Grant said a call for proposal has received 35 high-caliber applications. They’re currently being shortlisted through an external peer review process, and CEPI’s scientific advisory committee will reach a conclusion by the end of June.
The grant comes as Nigeria suffers an unprecedented Lassa fever outbreak. The overall fatality rate is 1%, but for this year, it has reached 22% among confirmed and probable cases in the current Nigerian outbreak, the WHO reported.
MERS, first identified in 2012, causes severe respiratory illness, and it resulted in 186 cases and 36 deaths during an outbreak in South Korea in 2015.
A partnership between Inovio and South Korea’s GeneOne, with help from the Walter Reed Army Institute of Research, currently has the most advanced MERS vaccine program in phase 1 testing.
As for Lassa, according to a comprehensive summary by CEPI, no vaccine has progressed out of preclinical stages. Inovio, for one, is working on a candidate with the U.S. Army Medical Research Institute for Infectious Diseases.
Under Themis’ second partnering agreement with the Coalition for Epidemic Preparedness Innovations (CEPI), the Vienna-based company is eglible to receive up to $21m to push Phase III testing of its Chikungunya vaccine candidate, MV-CHIK. CEPI’s Chikungunya-vaccine development mandate was launched in 2019 with support from the European Commission’s Horizon 2020 programme.
In Phase II trails, Themis’ live-attenuated, measles-vectored chikungunya vaccine (MV-CHIK), which has FDA fast track and EMA PRIME status, showed good safety and tolerability as well as immunogenicity. Themis announced that the non-dilutive funding will provide a significant portion of the capital required for Themis’ Phase III clinical trial of MV-CHIK expected to start this year. The pivotal multi-center Phase III trial will be launched in Europe, US and the Americas and will also test a single-shot regimen.
The World Health Organization (WHO) has highlighted Chikungunya, which causes arthritis-like symptoms, as a major public health risk. The disease was first identified in Tanzania in 1952, with sporadic outbreaks of the disease reported subsequently across Africa and Asia.In 2004 the disease began to spread quickly, causing large-scale outbreaks around the world. Climate change is set to further amplify the threat posed by Chikungunya. As the climate warms, more areas across the world will become habitable for the mosquito vectors that transmit the virus, thereby increasing the size of the human population at risk of infection. In 2007, for example, an outbreak of Chikungunya virus infections was declared for the first time in Europe, with more than 200 human cases reported in Italy.
Since the re-emergence of the virus, the total number of cases has been estimated at over 3.4 million in 43 countries.Chikungunya is spread by the bites of infected female Aedes mosquitoes and causes fever, severe joint pain, muscle pain, headache, nausea, fatigue and rash. Joint pain is often debilitating and can persist for weeks to years.
Themis’ first partnership with CEPI, announced in March 2018, provided up to $37m in funding to support vaccine development and manufacturing for Lassa fever and MERS.
Merck & Co. will partner with Themis Bioscience to develop vaccine candidates based on Themis’ measles virus vector-based platform, through the Austrian biotech’s first-ever collaboration with a major biopharma.
Themis says the platform, which it licenses from the Institut Pasteur in Paris, can incorporate large recombinant genes coding for selected antigens into its genome. Vaccines developed through the platform are designed to deliver multiple selected antigens—such as full-length proteins or virus-like particles—directly to macrophages and dendritic cells, thus triggering a specific immune response to the selected antigens.
The companies have committed to developing vaccine candidates against an undisclosed disease target. Speaking with GEN, Themis CEO Erich Tauber, MD, would not disclose what indications the companies are focusing on.
“What I can say is the measles virus vector technology allows to exploit infectious diseases and cancer indications, and when it comes to infectious diseases, we use the measles virus to bring our specific antigens into the body. Those might be difficult to express in normal cell systems,” Tauber said.
“Another advantage is we can use exactly the same manufacturing process for each new vaccine target. And when it comes to cancer, the measles virus itself has strong oncolytic activity,” Tauber said, such as mediating tumor cell lysis, T cell activation, and specific tumor cell targeting. “The measles virus can kill cancer cells. And we enhance this activity by putting in specific therapy enhancing proteins like immune modulators.”
Merck has agreed to make an unspecified equity investment in Vienna-based Themis under the companies’ collaboration and exclusive license agreement, which Themis said could generate for it more than $200 million.
In addition to the equity investment, Merck agreed to provide Themis an unspecified amount of research funding, as well as up to approximately $200 million in payments tied to achieving development and sales milestones, plus royalties on approved products from the collaboration.
“We continue to evaluate technologies with the potential to deliver novel vaccine candidates,” Daria Hazuda, PhD, CSO, Exploratory Science Center and vp of infectious diseases and vaccine discovery at Merck, said in a statement. “We look forward to collaborating with the scientists at Themis.”
Surging vaccines sales
Merck credits human vaccines, along with cancer treatments led by the blockbuster immunotherapy Keytruda® (pembrolizumab), with its most recent positive quarterly results.
During the second quarter, Merck said, overall company sales rose 12% year-over-year to $11.760 billion—or 15% excluding the effect of foreign exchange rates. The biopharma giant finished Q2 with $2.670 in GAAP net income, up 56% from $1.707 billion in the second quarter of 2018.
However, human health vaccines sales zoomed 33% year-over-year, to $2.0 billion, or 36% when excluding currency impacts. Two vaccines led Merck’s sales surge: PROQUAD (Measles, Mumps, Rubella and Varicella Virus Vaccine Live) and VARIVAX (Varicella Virus Vaccine Live), a vaccine to help prevent chickenpox, saw their combined sales jump 58% in Q2, to $675 million from $426 million in the year-ago quarter. Merck cited higher demand, including from private-sector buyers, and U.S. pricing, as well as government tenders in Latin America and higher demand in Europe.
Merck also reported a 46% quarterly sales jump (50% excluding currency impacts)—to $886 million from $608 million in Q2 2018—for the tandem of GARDASIL [Human Papillomavirus Quadrivalent (Types 6, 11, 16, and 18) Vaccine, Recombinant] and GARDASIL 9, vaccines indicated to treat some cancers and other diseases caused by HPV. Merck cited primarily public sector buying patterns, U.S. demand and pricing, and the ongoing commercial launch in China, as well as higher demand in Europe, driven primarily by increased vaccination rates for both boys and girls.
Expanding pipeline range
Merck’s collaboration with Themis is likely to expand the range of indications for vaccine candidates in the pipeline of Themis, which has focused most in developing vaccines against infectious diseases, but also has several preclinical immuno-oncology programs.
In October 2018, Themis signed an exclusive worldwide license agreement of undisclosed value with Max-Planck-Innovation GmbH, the technology transfer agency of the Max Planck Society, to develop, manufacture, and commercialize therapies based on an oncolytic measles virus platform jointly developed by the Eberhard-Karls-University Tübingen and the Max Planck Institute for Biochemistry.
Themis’ most advanced pipeline program is an unpartnered chikungunya vaccine candidate MV-CHIK, which Tauber said remains on track for a Phase III trial set to begin later this year. The pivotal multi-center trial will test a single-shot regimen, with patients to be dosed at centers in Europe, the U.S., and the Americas.
In June, Themis was awarded up to $21 million in non-dilutive capital toward development of the chikungunya vaccine by the Coalition for Epidemic Preparedness Innovations (CEPI), part of CEPI’s third call for proposals with support from the European Union’s (EU’s) Horizon 2020 research funding program under grant agreement No. 857934. The award is intended to accelerate regulatory approval of the chikungunya vaccine and ensure that at-risk populations have access to the vaccine by funding a “significant” portion of the capital needed for the Phase III trial, Themis said at the time.
Themis once planned to fund the Phase III trial through an up-to-€55.3 million ($61.3 million) initial public offering on Euronext Amsterdam, but postponed the IPO in November 2018, citing adverse market conditions.
The chikungunya candidate is also under study in a Phase II trial (NCT03807843) designed to assess the vaccine’s safety and immunogenicity in adults with a history of chikungunya infection; and another Phase II trial (NCT03635086) designed to investigate the immunogenicity, safety, and tolerability of MV-CHIK as well as the long-term durability of anti-chikungunya antibody response after administration of different dose levels of the vaccine in three different formulations.
Last year, CEPI awarded Themis up to $37.5 million toward developing vaccines for Lassa fever and Middle East respiratory syndrome (MERS).
Merck has struck a deal to buy Themis to accelerate the development of a COVID-19 vaccine. The takeover will see Merck, a latecomer to the response to SARS-CoV-2, apply its vaccine capabilities to a candidate based on Themis’ measles vector platform that is set to enter the clinic this year.
Themis is developing a pipeline of vaccines based on a measles virus vector platform it licensed from Institut Pasteur. By engineering the virus to express different antigens, Themis aims to use the same vector and manufacturing system to develop vaccines that induce protection against a wide range of infectious diseases, including COVID-19.
“Together with Institut Pasteur, we have worked on very closely related viruses like SARS and MERS [and] demonstrated the platform is very useful in eliciting an immune response,” Themis CEO Erich Tauber said. “We started [SARS-CoV-2] vector design in February. We have started in vivo models … and are now preparing for clinical trials.”
Merck is now set to apply its vaccine capabilities to the program. The Big Pharma has a major human vaccine operation, which generated sales of $8.4 billion last year, but it stayed on the sidelines in the early days of the pandemic as peers such as AstraZeneca, Pfizer and Sanofi placed bets on COVID-19 vaccine candidates.
News of a change in strategy came late in April when Merck said it was talking to “multiple groups” about three viral vector platforms. The talks manifested in an agreement to buy Themis, a privately owned Austrian biotech, for an undisclosed sum. In selecting Themis as a key plank of its COVID-19 strategy, Merck has indicated it thinks the biotech’s vaccine can clear a high bar.
“The task before us is one that requires a vaccine that will be quite stimulatory and that will yield neutralizing antibodies ideally with a single immunization. Of course, it must first be safe because you’re talking about a vaccine that would in principle be given to much of the world’s population,” Roger Perlmutter, president of Merck Research Laboratories, told investors last month.
Themis, as part of a consortium featuring Institut Pasteur, partnered with the Coalition for Epidemic Preparedness Innovations (CEPI) to develop a COVID-19 vaccine in mid-March. Earlier this month, Themis disclosed a deal with service provider ABL Europe covering the production of the vaccine in France.
Merck, which plans to start testing the vaccine in humans this year, has previously said it is trying to identify internal resources and contract manufacturers that can enable it to produce 1 billion doses of a COVID-19 vaccine and plans to make Themis’ shot at sites in the U.S. and Europe. The ability of Merck to bring such scale to bear factored into Themis’ decision to sell up.
“The limiting step for everybody will be manufacturability and manufacturing capacity. Merck brings an enormous skill level, expertise and capacity in terms of manufacturing technology. They’re using very similar technology already. They have been manufacturing measles vaccines for 60 years or so,” Tauber said.
Merck bought into the concept behind Themis’ platform last summer when it tasked the biotech with developing vaccines against an undisclosed target and invested in its series C round. Now, Merck has decided to acquire its partner outright.
The takeover will give Merck ownership of the platform, which Themis thinks has immuno-oncology applications, and a pipeline led by a phase 3-ready chikungunya vaccine candidate. Merck also sees the acquisition as a way to “ build a pandemic preparedness capability” against future threats.
For now though, COVID-19 is the focus. Merck and CEPI have entered into a memorandum of understanding about the need to make the vaccine “accessible to those who need it, including low-income, middle-income and high-income countries, based on the medical need.” Tauber raised Merck’s approach to vaccine access in explaining why it is the right partner, citing the Big Pharma’s work on Ebola as evidence that it is “very enthusiastic about global supply of vaccines.”
After Merck & Co. got off to a late start in the COVID-19 vaccine race and made an early exit, the drug giant is in talks to aid the global vaccine manufacturing effort.
The drugmaker is “actively involved” in discussions with governments, health agencies and other pharmaceutical companies to “identify the areas of pandemic response where we can play a role, including potential support for production of authorized vaccines,” a spokesman said via email.
News of the talks comes about two weeks after Merck abandoned both its coronavirus vaccine candidates—one it acquired through its Themis buyout and the other it was studying in partnership with IAVI. Merck said the two shots had produced immune responses weaker than those prompted by natural infections as well as by other COVID-19 vaccines.
Still, the company believes it has an “important responsibility to contribute to the pandemic response,” the spokesman said, and remains “at the ready to do so.”
While Merck hasn’t indicated which companies it could help with production, there has been industry talk about a potential tie-up with Novavax. After the vaccine biotech last month presented positive phase 3 data on its candidate, Evercore ISI analyst Josh Schimmer said he suspected Merck might “step up” as Novavax’s manufacturing partner.
Novavax CEO Stan Erck then told CNBC’s Meg Tirrell that Merck “could be a good partner for us as they don’t have a competing product.” He also named GSK as a company with those capabilities. At the time, Merck told Tirrell it was focused on therapeutics.
Meanwhile, Merck has two coronavirus therapeutics in development—MK-4482 and MK-7710—and the company believes it can make a “meaningful contribution” to the fight against the pandemic by focusing its resources on those candidates, its spokesman said.
Last summer, as COVID-19 vaccine programs raced forward, Merck CEO Ken Frazier said the hype about vaccines launching in late 2020 was doing a “grave disservice” to the pandemic fight. Vaccines previously took years to develop, he pointed out, and Merck itself was responsible for many of them.
He wasn’t alone. Merck and other major vaccine players were taking a slower, time-tested approach, experts said, but their vaccines could end up reaching more people worldwide than more revolutionary shots would. Things didn’t turn out that way. Pfizer, Moderna, AstraZeneca and other programs are now either rolling out or nearing rollouts, while several leading vaccine giants have either exited the field or faced R&D setbacks.
If Merck does strike a manufacturing deal with a COVID-19 vaccine player, it won’t be the first company to do so. After an R&D setback on its GSK-partnered vaccine, Sanofi last month said it would produce 100 million doses of the Pfizer-BioNTech mRNA vaccine for Europe.
The Pfizer-BioNTech team has also enlisted Swiss drugmaker Novartis in its global push to produce billions of doses. In a deal unveiled in late January, Novartis said it would allow BioNTech access to its site in Stein, Switzerland. Manufacturing there will start next quarter, and doses will be ready from the site by the third quarter.
Also this week, Teva said it was in talks to help with COVID-19 vaccine production. The company has sites in Israel, Europe and the U.S. that could be used in the global effort, CEO Kåre Schultz said, according to The Wall Street Journal.
(Picture: Rudolf Jaenisch & Richard A. Young, Whitehead Institute)
안녕하세요 보스턴 임박사입니다.
Omega Therapeutics는 2015년에 Whitehead Institute의 Rudolf Jaenisch 교수와 Richard A. Young 교수에 의해 Cell Stem Cell에 발표한 Insulated Genomic Domains (IGDs)를 이용해서 Epigenomic Programming을 할 수 있다는 아이디어로 2017년에 Flagship Pioneering의 David Barry 박사에 의해 설립되었습니다.
CAMBRIDGE, Mass. (December 10, 2015) –Whitehead Institute researchers have created a map of the DNA loops that comprise the three dimensional (3D) structure of the human genome and regulate gene expression in human embryonic stem (ES) cells and adult cells. The location of genes and regulatory elements within this chromosomal framework could help scientists better navigate their genomic research, establishing relationships between mutations and disease development.
“This is transformational,” says Whitehead Member Richard Young. “This map allows us to predict how genes are regulated in normal cells, and how they are misregulated in disease, with far greater accuracy than before.”
In order to regulate gene expression, a regulatory element needs to contact its target gene. Through looping, element/gene partners that are distant from each other in linear DNA can be brought together. Most disease mutations occur in regulatory elements, but if the partnership between a seemingly far-flung gene and the regulatory element is not known, the mutation data is of limited use. This draft map, which can help scientists predict the relationships between mutated elements and their target genes, is described online this week in the journal Cell Stem Cell.
“When thinking about disease, we need to think about the structure of the genome in 3D space because that is how we now understand that genes are regulated,” says Xiong Ji, a postdoctoral researcher in the Young lab and a co-author of the Cell Stem Cell paper.
One of Ji’s co-authors, graduate student Daniel Dadon, agrees. “This three-dimensional information helps us to interpret regulatory and mutational data with unprecedented accuracy. It’s not just a bag of genes and regulatory elements in the nucleus—this is a highly organized structure that confers function.”
Previous research in mouse ES cells by Young’s lab and others determined that a chromosome’s DNA is formed into loops that are anchored at their bases by proteins called CTCFs. The benefits of the loops are two-fold. First, the loops help organize and package two meters of DNA to fit into a nucleus that is approximately 5 micrometers in diameter. Second, each loop creates an insulated neighborhood that restricts the action of a regulatory element to genes that resides in the same loop. As graduate student and co-author Diego Borges-Rivera states, “The genome’s 3D shape is a key mechanism underlying gene regulation.”
By studying human ES cells, scientists in the Young lab and the lab of Whitehead Founding Member Rudolf Jaenisch created an initial genome map consisting of 13,000 loops established by CTCF anchors and determined that the average insulated neighborhood is 200 kb in length and contains a single gene. The team found that most of the the mapped CTCF anchor sites in the human ES cells genome are maintained in other human cell types and furthermore, that these loop anchor sequences are highly conserved in primate genomes. Such a surprising degree of conservation indicates that these neighborhoods create a foundational framework for gene regulation that is maintained throughout development and across species.
In a further finding that underscores the importance of the genome’s 3D structure in human health, the Whitehead team found that the CTCF anchor regions are mutated in a broad spectrum of cancer cells. The team predicts that these new maps of the human genome will provide the foundation for improved understanding of the genetic alterations that cause many additional diseases.
This work was supported by the National Institutes of Health (NIH grants HG002668 and HD 045022), National Cancer Institute (NCI), the Erwin Schroedinger Fellowship (J3490) from the Austrian Science Fund, and the Simons Foundation (SFLIFE 286977). Jaenisch is a founder of Fate Therapeutics and Young is a founder of Syros Pharmaceuticals.
2017년 7월부터 2019년 6월까지 우선주 투자방식으로 $28 Million Series A를 받았습니다.
Series A Preferred Stock Financing. From August 2017 to June 2019, we issued and sold to investors in private placements an aggregate of 56,775,232 shares of our Series A preferred stock at a purchase price of $0.50 per share, for aggregate consideration of approximately $28.4 million.
그리고 2019년 9월에 처음으로 회사의 존재를 세상에 알렸습니다. 2년여의 Stealth mode를 거친 후 발표를 한 것이죠. 이 당시에는 모든 프로그램이 전임상 단계였습니다.
Flagship Pioneering launched Omega Therapeutics, a company aiming to take genomic medicine “to the next level.” Founded on the work of two MIT professors, the company is working on treatments that adjust gene expression up or down without making permanent changes to the genome.
Richard Young, Ph.D., and Rudolf Jaenisch, M.D., first described how 3D closed loops of DNA control genomic activity in 2015. Long strands of DNA make these loops because they have to fit into the cell’s nucleus—the loops help “organize and package two meters of DNA” to fit into a space that is about 5 micrometers, or five millionths of a meter, across, the researchers said in a statement at the time. Each loop is an “insulated neighborhood” of one or more genes and their regulatory elements.
Omega is targeting these neighborhoods, called Insulated Genomic Domains (IGDs), with a platform that could be applied to a variety of ailments.
“If you think about it, other than some viral and other infections, pretty much all human disease is due to the dysregulation of genomic expression,” Omega CEO Mahesh Karande, a Novartis alum and former CEO of Macrolide, told FierceBiotech. “Disease mostly occurs because of dysregulation of the genome, by genes not being expressed at the right level. They’re over- or under-expressed. We are able to tune that expression to the native level it’s supposed to be at.”
The Cambridge, Massachusetts-based biotech is mapping IGDs to different diseases and figuring out which of these neighborhoods plays a role in different diseases. From there, it will create treatments it calls Omega Controllers that adjust gene expression to healthy levels.
This adjustment happens without making permanent changes to the genome by switching genes on or off, cutting disease-causing genes out or putting in a healthy version of a faulty gene.
“In nature, generally things are not all the way on or all the way off, but rather turned to a very specific range in a healthy setting,” said Omega Chief Scientific Officer Thomas McCauley, Ph.D., the former CSO of Macrolide and Translate Bio. “Our Omega Controllers are able to target IGDs using the map that Mahesh mentioned and target the right place on that IGD to restore gene function at the right level.”
Because the approach could work for so many disease areas, Omega plans to ink some partnerships as well as work on its own pipeline, said David Berry, M.D., Ph.D., a general partner at Flagship, in a statement. Omega’s treatments could be used to boost the efficacy of in vivo and ex vivo therapies, he said.
In the in vivo space, checkpoint inhibitors are a potential candidate.
“Many of them are not as effective as you’d like them to be. Sometimes immuno-oncology agents act only on 30% of the patient population,” Karande said.
Omega could identify specific genes in patients that affect how they respond to these treatments. For example, if a gene is expressed in patients who don’t respond to a certain immuno-oncology drug, Omega might knock down the expression of that gene to make that drug more effective. On the ex vivo side, Karande envisions Omega’s technology being used when cell therapies are being engineered outside the body.
이듬해인 2020년에 $85 Million Series B를 하면서 임상진입을 시도한다고 발표를 했습니다.
Less than a year after launch, Omega Therapeutics is getting an $85 million cash boost. It will push a pipeline of treatments toward the clinic as well as bankroll the identification of new targets for genomic medicines.
“We had founded Omega with a long-term vision to create a controllable epigenomic programming platform,” Omega CEO Mahesh Karande told Fierce Biotech. Rather than switching genes on and off, cutting out disease-causing genes or replacing them with healthy versions, Omega’s platform is designed to adjust gene expression to healthy levels.
The company’s work is based on “neighborhoods” of genes and their regulatory elements found in loops of DNA called Insulated Genomic Domains (IGDs). These loops occur because long strands of DNA need to fit into the cell’s nucleus.
“In nature, generally things are not all the way on or all the way off, but rather turned to a very specific range in a healthy setting,” Omega Chief Scientific Officer Thomas McCauley, Ph.D., said in a previous interview. Omega’s “epigenomic controllers” are designed to target the right place on specific IGDs to restore gene function at the right level, he said.
Since launch, Omega has been working to figure out which neighborhoods play a role in different diseases.
“We could have gone in various directions,” Karande said. But Omega landed on a handful of areas. It’s advancing five programs spanning oncology and inflammation as well as autoimmune, metabolic and rare genetic diseases, the first of which should hit the clinic in 2021.
In addition to tweaking gene expression without making permanent changes to the genome, Omega’s approach offers advantages over a small-molecule approach to epigenetics.
“There are a number of companies developing small-molecule therapies for epigenetic targets, almost exclusively in cancer,” McCauley.
“The issue is really specificity, in having those molecules go everywhere in the body as opposed to having them go to specific cell types and specific locations in the genome,” McCauley continued, adding that the benefits of such treatments might outweigh the risks in oncology but that this risk-benefit profile may be unacceptable in other diseases.
In its first efforts, Omega is going after targets with links to specific diseases that are well understood, McCauley said. Moving forward, it will take advantage of the lessons it learns to look for new targets.
“We’re looking for the ability to expand laterally,” he added.
One of those lateral expansions could be into COVID-19. Since inflammation plays a big role in COVID-19 infection, Omega could leverage the work it’s already done in that space to quickly move into drug development against the new coronavirus.
Right now, it’s all systems go with its five—potentially six—programs. Karande said the company would be “remiss” if it did not ink partnerships.
“We are absolutely open to partnering with people. We have a robust discovery platform that has many, many more targets in the pipeline, so yes, partnering is definitely in the cards for us,” he said.
그리고 2021년에는 $126 Million 펀딩을 통해서 OTX-2002 (HCC) 약물의 임상진입과 다른 전임상 약물의 개발을 발표했습니다.
Omega Therapeutics is taking it up a notch. The “genome-tuning” biotech raised $126 million to get its lead program, a treatment for liver cancer, into the clinic as well as to advance a clutch of other preclinical prospects including a treatment for acute respiratory distress syndrome (ARDS), a life-threatening lung injury that can result from COVID-19 infection.
The company is working on a new class of treatments called “epigenomic controllers,” which are designed to adjust the expression of target genes. Unlike gene therapies and gene editing approaches that switch genes on and off, cut out disease-causing genes or replace faulty genes with their healthy versions, Omega’s treatments tune gene expression up or down without making permanent changes to DNA.
Its work is based on “neighborhoods” of genes and their regulatory elements found in loops of DNA called Insulated Genomic Domains (IGDs). These loops occur because long strands of DNA need to fit into the cell’s nucleus. Omega’s “epigenomic controllers” are designed to restore gene function to healthy levels by targeting the right place on specific IGDs.
The series C financing follows an $85 million B round in July 2020, which went toward identifying new targets and driving several programs across multiple disease areas through preclinical development.
“When we started, we needed to explore the depth of the platform—we didn’t want to pigeonhole ourselves,” said CEO Mahesh Karande. “That’s how we delineated eight programs at five different targets.”
Omega has started IND-enabling studies for its lead program, OTX-2002, an epigenetic controller programmed to control expression of c-myc, an elusive cancer-driving gene. It is developing the treatment for hepatocellular carcinoma, the most common form of liver cancer.
Unlike Gilead’s antiviral Veklury (remdesivir) and anti-SARS-CoV-2 antibodies from Eli Lilly and Regeneron, Omega’s COVID-19 treatment focuses on ARDS, which is caused by an inflammatory response in the lungs.
“We treat diseases created by functional or structural changes in IGDs and ARDS creates a functional change in a multigenic IGD where cytokines get supercharged and expressed,” Karande said. With its epigenomic controller, Omega aims to reduce the expression of those cytokines.
The company hopes the treatment will fill a gap in COVID-19 treatment.
“The standard of care [for ARDS] is quite insubstantial and largely palliative, involving mechanical ventilation and where possible, steroids,” said Omega Chief Scientific Officer Thomas McCauley, Ph.D.
Besides liver cancer and ARDS linked to COVID-19, Omega is focusing on regenerative medicine, inflammatory diseases, alopecia, non-small cell lung cancer and a group of skin conditions called neutrophilic dermatoses.
The financing will also bankroll a manufacturing scale-up as well as the expansion of Omega’s technology. As it develops drug candidates, it will continue to improve its platform, “taking the guesswork out of it” and making sure it can keep generating new drugs in a reliable and replicable way, Karande said.
As it continues its quest toward the clinic, Omega will build up its workforce, particularly its clinical organization and manufacturing unit.
Omega Therapeutics, Inc. (Nasdaq: OMGA) (“Omega”), a development-stage biotechnology company leveraging its OMEGA Epigenomic Programming™ platform to harness the power of epigenetics to develop a new class of DNA-sequence-targeting, mRNA-encoded programmable epigenetic medicines, today announced the pricing of its initial public offering of 7,400,000 shares of its common stock at a price to the public of $17.00 per share. All of the shares of common stock are being offered by Omega. The gross proceeds from the offering, before deducting underwriting discounts and commissions and estimated offering expenses payable by Omega, are expected to be approximately $125.8 million, excluding any exercise of the underwriters’ option to purchase additional shares. Omega’s common stock is expected to begin trading on the Nasdaq Global Select Market under the ticker symbol “OMGA” on July 30, 2021. The offering is expected to close on August 3, 2021, subject to satisfaction of customary closing conditions. In addition, Omega has granted the underwriters a 30-day option to purchase up to an additional 1,110,000 shares of common stock at the initial public offering price less underwriting discounts and commissions.
Goldman Sachs & Co. LLC, Jefferies LLC and Piper Sandler are acting as joint book-running managers of the offering. Wedbush PacGrow is acting as lead manager.
2023년에 OTX-2002의 8명의 환자에 대한 Preliminary clinical trials 결과를 발표했는데 Genetic selectivity와 MYC 가 dose-range에 맞게 감소하는 것을 보고함으로써 플랫폼의 기술이 임상에서도 작용을 한다는 것을 일단 소규모 임상에서 증명을 했습니다.
The investigational mRNA therapeutic OTX-2002 demonstrated encouraging safety, tolerability, and pharmacokinetics in a small population of patients with hepatocellular carcinoma (HCC) and other solid tumors related to the c-MYC (MYC) oncogene, according to a press release on findings from the phase 1/2 MYCHELANGELO I trial (NCT05497453).1
Investigators reported highly specific on-target engagement and epigenetic changes among all 8 patients receiving 0.02 mg/kg (n = 4) or 0.05 mg/kg of OTX-2002 (n = 4) every 2 weeks. The agent’s modulation of MYC rapidly and durably downregulated MYC oncogene expression in all 8 patients; investigators highlighted a mean reduction of 55% at 7 days following treatment.
Treatment with OTX-2002 also produced consistent pharmacokinetic data at both dosing levels, as investigators observed little variability and quick clearance within patients. Additionally, there was no accumulation after additional doses of OTX-2002, and the agent produced low and transient levels of immune response with no adverse effects (AEs) impacting pharmacokinetics. Investigators reported that both initial dose levels of the agent demonstrated anti-tumor activity below the predicted threshold established by preclinical models.
OTX-2002 was well tolerated among patients, and investigators observed no dose-limiting toxicities. Patients mostly experienced grade 1 or 2 AEs, the most common of which included infusion-related reactions (26%) such as fever and chills. These toxicities appeared to be comparable with the known profiles of other FDA-approved agents administered via lipid nanoparticles.
“We are thrilled to see that all 8 patients evaluated at these initial low doses demonstrated clear evidence of on-target epigenetic changes and correlated rapid, robust and durable decreases in MYC mRNA expression levels,” Thomas McCauley, PhD, chief scientific officer at Omega Therapeutics, said in the press release. “These early clinical data are consistent with our preclinical experiments, giving us confidence that our approach has the potential to translate to anti-tumor activity and clinical benefit. Coupled with encouraging safety and predictable pharmacokinetics, we believe that OTX-2002 holds transformative potential for patients living with HCC.”
In the ongoing open-label MYCHELANGELO I trial, investigators are assessing the safety, tolerability, pharmacokinetics, and preliminary anti-tumor activity of OTX-2002 on its own in part 1 and with standard-of-care treatments in part 2 among those with relapsed/refractory HCC and other solid tumor types associated with MYC oncogene expression. Investigators are conducting the trial at clinical sites in the United States and Asia. As of the data cutoff point of September 18, 2023, a single patient with HCC remained on treatment in the 0.05 mg/kg cohort.
The trial’s primary end points are dose-limiting toxicities, treatment-emergent AEs, overall response rate, and duration of response.
Patients 18 years and older with metastatic, advanced, or recurrent solid tumors that have progressed following standard-of-care therapy and intermediate or advanced stage, Child-Pugh A HCC not amenable to locoregional therapy or curative treatment approaches are able to enroll on the study. Having an ECOG performance status of 0 or 1 is another requirement for enrollment.
“We look forward to continuing to work with clinical investigators, patients, and the FDA as we advance our MYCHELANGELO clinical program and evaluate the potential of OTX-2002 to bring a new treatment option to the community [of patients with liver cancer],” Mahesh Karande, president and chief executive officer at Omega Therapeutics, said in a press release at the time of the orphan drug designation.2
References
Omega Therapeutics announces promising preliminary clinical data for OTX-2002 from ongoing MYCHELANGELO™ I trial. News release. Omega Therapeutics. September 26, 2023. Accessed September 26, 2023. https://shorturl.at/hmqNZ
Omega Therapeutics receives orphan drug designation for OTX-2002 for the treatment of hepatocellular carcinoma. News release. Omega Therapeutics. November 2, 2022. Accessed September 26, 2023. http://bit.ly/3fqiafd
금년 새해 첫날에 Flagship Pioneering과 Novo Nordisk의 계약에 의해서 Omega Therapeutics의 IGD Platform을 이용해서 Obesity 치료제를 개발하는 공동계약을 맺었고 계약 규모는 $532 Million입니다.
As part of a pact with Flagship Pioneering, Novo Nordisk has inked separate cardiometabolic disease research deals with two Flagship-founded biotechs that are worth up to $532 million each.
The freshly formed agreements are with Omega Therapeutics and Cellarity, two Massachusetts biotechs that fall under Flagship’s umbrella. The partnerships are part of a broader ecosystem collaboration Flagship and Novo Nordisk’s Bio Innovation Hub struck up in mid-2022 that aims to quickly build a portfolio of breakthrough medicines for cardiometabolic and rare diseases.
Using Novo’s disease expertise and technology from Flagship’s bioplatform companies, the goal is to generate three to five research programs within the first three years of the partnership, Novo Nordisk’s head of the Bio Innovation Hub Uli Stilz, Ph.D., told Fierce Biotech in an interview.
For these first two deals, each company, Novo Nordisk and Flagship’s Pioneering Medicines—an initiative that develops treatments by using and expanding Flagship innovations—will work together to advance their respective programs through preclinical development. Novo will then have the chance to take the programs into the clinic.
The Big Pharma will reimburse R&D costs and give each company and Pioneering Medicines the chance to make up to $532 million dollars in upfront and milestone payments, plus tiered royalties.
With Omega, Novo will look to expand upon its blockbuster obesity franchise (does the drug Ozempic ring a bell?) with the biotech’s platform, which is made to design programmable epigenomic mRNA medicines that precisely target and modulate gene expression at the pre-transcriptional level.
“We have been pioneers over a 20-year journey in obesity and we want to continue to be a pioneer,” Stilz said, adding that being a pioneer means entering uncharted scientific territory, which is where Omega comes in.
The biotech will use its platform technology to develop an epigenomic controller as part of a new obesity management approach. While many existing therapeutics for weight management focus on appetite regulation, Omega wants to target thermogenesis, a natural metabolic function that regulates overall energy balance.
“What is so exciting for us is that it’s a very different approach than what we have been doing so far,” Stilz said. “I haven’t seen something similar anywhere else. So, we’re really pushing the boundary of science and innovation through this co-creation and collaboration.”
Omega already has some proof-of-concept to support its mission to design programmable mRNA medicines by replicating how nature’s control system works, Omega President and CEO Mahesh Karande told Fierce Biotech. The company’s platform is applicable across almost every disease process, Karande said, and Omega is currently evaluating one of its assets in a phase 1/2 trial for patients with hepatocellular carcinoma.
Now, the company will put its platform to work to control metabolic activity and potentially develop a more durable approach to obesity management.
“Epigenomic control and epigenomic controllers have not existed before. We have literally created this field,” Karande said. “And now we have signed our first major agreement with a company that is an expert in obesity, metabolics and cardiovascular. So, for us, this is hugely validating.”
Meanwhile, Cellarity will plug away at creating a small molecule therapy to treat metabolic dysfunction-associated steatohepatitis (MASH)—the new term for nonalcoholic steatohepatitis (NASH)—a chronic and progressive liver disease for which there is no currently approved treatment. The indication has a high unmet patient need, with only four investigational treatments ever making it into phase 3 development for MASH, led by Madrigal Pharmaceutical’s resmetirom, which is awaiting an FDA decision this spring.
Cellarity and Novo hope to develop a small molecule therapy for the indication by using the biotech’s platform that is designed to provide new insight into cellular dysfunction and allow for drug creation that has been historically inaccessible using traditional drug discovery methods.
Cellarity combines biology, chemistry and AI machine learning to understand cell behavior, Novo’s Stilz explained. Novo Nordisk has previously connected with Cellarity, asking the biotech in September 2022 to identify novel cell behaviors involved in MASH disease progression, work that will now be expanded upon under the new research collaboration.
그리고 얼마전에 Omega Therapeutics는 현재 임상 중인 OTX-2002와 전임상 중인 몇개의 약물에 집중한다는 전략적 우선순위 결정을 하면서 35%의 인원 감축을 하는 구조조정안을 발표한 상황입니다. 일단 플랫폼의 초기 임상이 성공하는 것이 중요하기 때문에 올바른 결정을 한 것으로 보입니다. 좋은 결과가 있기를 바랍니다.
Omega Therapeutics, Inc. (Nasdaq: OMGA) (“Omega”), a clinical-stage biotechnology company pioneering the development of a new class of programmable epigenomic mRNA medicines, today announced financial results for the fourth quarter and full year ended December 31, 2023, and a strategic prioritization initiative to focus resources on near-term milestones to support long-term shareholder value.
“2023 was an important year for Omega where we executed to plan and demonstrated clinical validation of an epigenomic controller to regulate c-MYC in humans for the first time. These proof-of-platform clinical data, coupled with our research collaboration with Novo Nordisk in obesity, support the ability of the OMEGA platform to potentially address epigenomic regulation of almost all human genes across broad therapeutic areas including cancer, cardiometabolic conditions and liver regeneration,” said Mahesh Karande, President and Chief Executive Officer of Omega Therapeutics. “Initial clinical data from our ongoing Phase 1/2 MYCHELANGELO I trial of OTX-2002 demonstrated controlled modulation of MYC expression levels, one of the most challenging gene targets in oncology, and an encouraging disease control rate and stable disease in heavily pre-treated, late-stage HCC patients. We are within what we believe is a clinically meaningful dose range and, as we continue to see a promising safety profile for OTX-2002, have recently opened enrollment of Cohort 5. We look forward to sharing additional updates from this program throughout 2024.”
“Today we also announced a strategic prioritization, implemented to ensure we have sufficient resources to advance our lead program and maximize near- and long-term value creation from our platform. As part of this initiative, we are taking difficult but necessary actions to streamline our team and optimize our R&D efforts and cost structure to extend our cash runway into the first quarter of 2025. These changes will unfortunately affect a number of our colleagues, and we are grateful for their dedication and contributions to our mission,” continued Mr. Karande. “As we sharpen our focus, we look forward to the opportunities ahead to generate meaningful clinical data for OTX-2002, continue to demonstrate the broad potential of our platform, and establish additional partnerships. We remain steadfast in our mission to pioneer a new class of programmable epigenomic mRNA medicines to transform the treatment of a broad range of diseases.”
Recent Highlights and Key Anticipated Milestones
Development Pipeline and Platform
Advanced the Phase 1/2 MYCHELANGELO™ I clinical trial evaluating OTX-2002 in patients with hepatocellular carcinoma (HCC):
OTX-2002 continues to advance in monotherapy dose escalation.
As of March 24, 2024, data from the first three cohorts (0.02 mg/kg – 0.06 mg/kg) showed:
OTX-2002 continued to be generally well tolerated, with no dose-limiting toxicities observed.
Consistent dose-dependent pharmacokinetics with no drug accumulation observed following repeat doses.
All patients demonstrated controlled modulation and downregulation of MYC mRNA expression, an important oncogene regulating cell function and cell death.
The interim disease control rate (DCR) for the target population of HCC patients was 80%, reflecting 4 out of 5 efficacy-evaluable patients having a best overall response of stable disease. These patients had an average of three or more previous therapies and entered the trial with a life expectancy of less than 12 weeks. The DCR for patients with non-HCC solid tumors in the trial (n=5) was 40%, indicating the potential specificity of OTX-2002 for HCC.
The Company continues to evaluate patients with HCC in Cohort 4 at the 0.12 mg/kg dose level, which recently cleared the 28-day dose limiting toxicity (DLT) window. Based on preclinical experience and modeling, Omega believes this dose level is within the expected active dose range. In March 2024, the Company opened enrollment for Cohort 5 at a dose level of 0.3 mg/kg.
Omega expects to report additional updated clinical data from monotherapy dose escalation in mid-2024.
The Company plans for expansion into monotherapy and combination settings in mid-2024.
Announced research collaboration with Novo Nordisk to develop a novel therapeutic for obesity management:
The collaboration will leverage Novo Nordisk’s expertise in research and development within cardiometabolic diseases and Omega’s proprietary platform technology to develop an epigenomic controller designed to enhance metabolic activity.
Unlike traditional approaches focused on appetite suppression, the program aims to leverage precision epigenomic control to enhance thermogenesis, a naturally occurring metabolic process that burns calories.
Under the terms of the agreement, Novo Nordisk will reimburse all R&D costs and has the right to select one target to advance for clinical development. Omega and Flagship’s Pioneering Medicines are eligible to receive up to $532 million in upfront, development and commercial milestone payments, as well as tiered royalties on annual net sales of a licensed product, which will be split equally between the parties.
Continued to advance and expand OMEGA platform capabilities:
Presented new preclinical data supporting the breadth of Omega’s platform capabilities, including bidirectional and multiplexed epigenomic control of gene expression in liver inflammation and fibrosis at the American Association for the Study of Liver Diseases’ (AASLD) The Liver Meeting® 2023.
A HNF4A-targeting epigenomic controller led to a durable increase in HNF4α expression, preferential upregulation of HNF4α P1 promoter isoforms, and reduced key measures of fibrosis both in vitro and in vivo, supporting this development candidate’s potential for the treatment of fibrotic liver disease.
In preclinical models, liver-specific multiplexed targeting of CXCL9, CXCL10 and CXCL11 via an epigenomic controller led to a significant reduction in T-cell migration, a critical driver of inflammation-induced liver injury, supporting the potential of this approach as a novel treatment for inflammatory liver diseases.
Corporate
Announced cost reduction and strategic prioritization initiative to maximize near- and long-term value creation opportunities:
Following a strategic review, the Company has focused its pipeline and reduced overall headcount by approximately 35%. These fiscally disciplined actions are expected to extend the Company’s cash runway into Q1 2025.
Positions the Company to achieve key clinical data readouts from the monotherapy dose escalation and dose expansion stages of the MYCHELANGELO I clinical trial.
The Company will prioritize certain preclinical programs and platform efforts:
Prioritized preclinical programs include OTX-2101 for non-small cell lung cancer (NSCLC), the HNF4A program in liver regeneration, and development of an epigenomic controller for obesity in collaboration with Novo Nordisk.
Core work on platform biology, epigenomic controllers, and characterization of LNP delivery to the lung and other tissues will continue.
An updated corporate presentation is available on the Investors section of the Company’s website at https://ir.omegatherapeutics.com/.
Fourth Quarter and Full Year 2023 Financial Results
As of December 31, 2023, the Company had cash, cash equivalents and marketable securities totaling $73.4 million, which is expected to fund operations into Q1 2025.
Research and development (R&D) expenses for the fourth quarter of 2023 were $15.5 million, compared to $26.0 million for the fourth quarter of 2022. R&D expenses for 2023 were $77.2 million compared to $81.2 million in 2022. The $4.0 million decrease in R&D expenses in 2023 compared to 2022 was primarily due to lower external research and manufacturing costs, consulting and professional fees, and lab expenses, partially offset by an increase in personnel-related expenses, including stock-based compensation to support business growth, and facilities and other costs.
General and administrative (G&A) expenses for the fourth quarter of 2023 were $6.2 million, compared to $5.7 million for the fourth quarter of 2022. G&A expenses for 2023 were $26.2 million, compared to $23.7 million in 2022. The $2.5 million increase in G&A expenses in 2023 compared to 2022 was primarily due to higher professional and consulting fees, and facilities and other administrative costs.
Net loss for the fourth quarter of 2023 was $20.2 million, compared to $30.8 million for the fourth quarter of 2022. Net loss for the year ended December 31, 2023, was $97.4 million, compared to a net loss of $102.7 million for the year ended December 31, 2022. The decrease in net loss for 2023 compared to 2022 was primarily due to decreases in R&D expenses.
신중년의 정의는 50-65세 혹은 60-75세 등 기준이 다양하지만 무엇인가 일을 해야한다는 것은 동일합니다. 신중년이 할 수 있는 다양한 일 – 재취업, 창업, 사회공헌, 귀농/귀촌 등 – 에 대해 비교적 상세히 그리고 나름의 깊이와 객관성을 가진 자료가 있어서 이것을 읽으며 제가 느낀 부분을 아래에 발췌합니다. 310쪽의 방대한 자료에 대해 각 페이지와 글을 남깁니다.
P43. 퇴직 후 긍정적 반응을 보인 퇴직자는 대부분 경제적 여유가 있는 경우에 해당한다.
P46. 남성 신중년 중에서도 사회적 역할 규범을 내면화하지 않고 가정에서 자신의 역할과 공간을 만들면서퇴직에 대한 긍정적 정서와 반응을 보이는 경우도 있다.
P47. 무엇보다도 주된 일자리 퇴직 후 신중년에게 의미 있게 나타나는 변화는 스스로 할 일을 만들며 하루의 일상을 알차게 만들려는 모습이다. 회사에 다닐 때보다 기상 시간이 한 시간 정도 늦춰진 차이밖에 없다는 인식을 보이면서 집이라는 공간을 회사에서 일하는 공간처럼 만들어 무언가를 계속할 수 있는 분위기 및 여건을 조성했다.
p51. 구직 기간이 길어지기 시작한 공백 시기에 자연에서 새 사진을 찍는 취미를 갖게 되었다. 이러한 활동은 재직 중에 취미로 시작한 것이었는데 지금은 일상의 중요한 활동이 되었다. 처음에는 가족들이 모델이 되어 주었지만 1년이 지나자 녹록지 않아 새에 대한 사진을 찍기 시작했다.
p52. 취미활동으로 기타동호회에 나가고 있다.
p52. 경제적 안정성을 어느 정도 확보한 신중년일수록 자신이 하고 싶었던 일 혹은 전문성을 발휘하여 보람 있게 할 수 있는 일을 찾고자 하는 욕구를 쉽게 찾아 볼 수 있다.
p72. 기술직 의 경우 다른 직종에 비해 상대적으로 재취업이 용이했고 재취업 일자리 적응도와 만족도가 높은 편인 것으로 나타났다.
p73. 재취업한 일자리가 퇴직 후 별다른 준비 없이 우연히 지인 소개 등을 통해 처음으로 재취업한 경우에는 그 만족도나 적응도가 더욱 낮은 것으로 나타나기도 했다. 다른 일자리를 경험한 사례자들의 경우 현재 의 일자리에 높은 만족도를 보였다.
p74. 이직 경험이 있는 경우 재취업을 돕는 다양한 고용 서비스의 필요성을 절실하게 느꼈음을 토로했으며, 활용한 서비스들의 효과에 매우 만족하는 모습을 보였다. 특히 실제로 상담자를 만나서 받는 일대일 컨설팅의 효과를 매우 높게 평가하고 있었다.
p77. 구직 실패를 경험한다는 것은 그 과정에서의 다양한 구직 활동 경험이 누적되는 것을 의미하는 것이기도 하다. 이들 역시 구직 과정에서 좀 더 나은 일자리로의 진출을 위해 직업교육 및 훈련, 구직 기술 강화 교육 등의 공공서비스 활용을 시도했다.
p78. 관점을 바꾸면 새로운 길이 보이듯, 사례자들은 하향 이동된 경력 무관 일자리도 긍정적으로 바라보고자 노력하고 있었다. 이들 역시 이동 초기에는 하향된 일자리에 쉽게 적응하지 못하고 좌절감을 경험했으나 새간이 지남에 따라 겸허히 현실을 수용하고 적응하고자 노력했다
p79. 경력 무관 일자리를 경험하는 사례자 중에는 자포자기하는 심정으로 받아들였던 일자리에서 생각지 못했던 비전을 발견하는 경우도 있었다. 그러나 이러한 비전을 발견하게 되었던 것은 일자리와 상관없이 어디서든 최선을 다하고자 하는 자세가 있었기에 가능한 것이기도 했다.
p83. “완전히 새로운 분야로 직종을 바꿨으니까 이제 초보 아닙니까. 가능하면 계속해서 더 업무를 수행할 수 있도록 도전하고, 이론과 기술을 모두 겸비한 ‘현장의 기술자’라는소리를 듣고 싶어요. 그래서 산업기사 자격도 따고, 전기기사도 따고 싶죠. 이건 출세하고는상관없어요. 그저 제 업무 분야에서 열심히 하는 거지만, 개인적인 삶의 만족은 물론 사회에도 작게나마 기여하고 싶죠.” (배기섭, 60대)
p83. 새 경력 재취업자들의 경우에는 다른 유형에 비해서 구직 기술 향상, 구직 정보 검색, 직업훈련 등 구직을 위한 다양한 활동을 보다 자발적이고 적극적으로 수행하는 것으로 나타났다. 새 경력으로의 진입을 위해서 교육, 훈련, 자격 취득 등 상대적으로 많은 시간과 노력을 들였으며 이 과정을 기꺼이 받아들이며 최선을 다하는 모습을 보여 주었다.
p91. 소확행. 내가 좋아하는 일을 하는 거 자체가 소소하지만 행복한 거 아닌가요? 내 일이 있고 직업이 있다는 것, 내가 직접 벌어서 내 용돈을 쓴다는 것, 애들한테 기대지 않고 사는 것.
p91-96. 은행 33년 퇴직 후 시설 관리 기술자로 전환 한 배기섭 (가명) 예 – 경제적 자유는 있었으나 일의 의미가 중요했던 케이스.
p97. “인텔리라는 사람들이 흔히 빠지기 쉬운 게 선민의식이잖아요. 저도 그런 게 있었죠. 그런데 퇴직하고 전기기능사 자격 따려고 직업훈련학교에 다닐 때, 처음으로 지하철 첫차를 타 봤어요. 새벽 5시 반에 지하철이 꽉 차요. 건설 현장 근로자, 일용직 근로자, 주야간 교대 근무하는 사람들 등 정말 다양하죠. 그 현장에 있어 보니 이전에 모르던 새로운 세상이 드디어 보이더라고요.”
p98. 상대적으로 충분한 퇴직 준비 기간을 가진 신중년들의 경우 혹독한 고용 시장의 현실을 미리 파악하고, 재취업, 귀농·귀어·귀촌, 창업 등 퇴직 이후의 다양한 경로를 미리 탐색하여 퇴직에 따른 변화와 충격을 완화할 수있었다. 또한 보다 이르게 시작한 은퇴 준비는 주된 일자리에서의 경력 마무리 시간을 다음 경로를 위한 의미 있는 시간으로 활용할 수 있게 했다.
p99. 다양한 사례자들의 경우 은퇴에 대한 이른 준비와 긍정적이고 겸허한 수용의 태도를 보임으로써 어려운 시기를 극복해 가고 있었다.
p100-104. 제약회사 인사임원에서 방역회사 3년 후 파견단순업무 – 고재식 (가명)님 사례 – 긍정적으로 받아들이는 자세가 중요.
p106. 재취업에서의 자기 이해와 관련하여 한 가지 함께 고려할 것은 자신의 특성이 무엇인가를 아는 것뿐 아니라, 자신의 객관적인 위치를 확인해야 한다는 점이다.
p108. 취업을 준비함에 있어 내가 진입하려는 시장에 대한 이해는 기본이다. 재취업 역시 마찬가지다. 그런데 재취업자들의 경우에 ‘고용 시장’에 오래 있었다는 사실만으로 자신이 현재 고용 시장에 대해서 ‘잘 알고 있다’는 편향에 빠져 실제 상황을 이해하는 데 소홀한 모습을 보이는 경우가 많다.
p108. “스마트공장추진단 사업은 정부에서 최소 10년을 내다보고 계획을 짠 거예요. 현재 민간 주도로 가고 있는데 지금 그 고도화 단계에서 진정한 스마트 공장은 2035년 정도에 중소기업에서 나오기 시작할 거라고 예측하고 있어요. 모든 정보가 자동화되고 그에 따라서 로봇이 움직이고 하는 그런 모델이 2035년 정도에 나올 거라는 거죠. 그럼 앞으로 10년 이상 있어야 하는 거잖아요. 제가 지금 53세인데 10년 후면 63세일 거 아니에요. 저는 최소한 그때까지는 일을 할 거라고 생각해요. 그게 희망이죠.”
p118. 재취업 준비 시 중요한 것은 페달을 빨리 밟는 것이 아닌 오래 밟을 수 있는가의 여부이다. 조바심을 내지 말고 장기적으로 준비하고 싶은 일이 있다면 시간을 좀 더 할애해서라도 그 일에 도전해 볼 것을 권유했다. 경우에 따라서 원하는 경력 목표를 달성하기까지 너무 많은 시간이 소요된다면, 비교적 단순 직종의 업무로 생계를 유지하며 훈련을 병행하는 것도 방법임을 조언해 주고 있었다. 특히 새 경력으로 진출을 도모하는 경우에 그쪽으로 진입하기 위해 상대적으로 많은 준비와 시간을 할애해야 하는 경우들이 있을 것이다.
p119, “포기하지 말아야 해요. 지금 좌절하고 실패하는 이 상황이 도약을 위한 기회일 수 있거든요. 재취업이 어렵다고 해서 인생 헛살았다는 생각을 하면 안 돼요. 우리 인생이라는 게 자전거를 타고 계속 나아가는 과정이거든요. 근데 빨리 갈 필요가 뭐가 있어요? 굳이 속도 내지 않고 천천히 가도 돼요. 중요한 건 페달에서 발을 떼지 않는 거거든요. 페달 놓는 순간 쓰러지잖아요. 주위에 사람이 있고, 많은 제도가 있으니 포기하지 않고 페달을 계속 밟는 거, 그게 제일 중요해요..”
“조급해하지 말자. 돌아가더라도 제 길로 가면 된다. 계속 속으로 되뇌었어요. 포기하지 않고 처음 마음대로 계속 취업에 도전해 보기로 결심했죠. 욕심은 소용이 없어요. 그렇다고 내가 가진 경험까지 버리고 싶지 않았어요. 어디든 내가 가진 노하우를 응용할 수 있고 접목시킬 수 있는 일을 찾아보고 싶었어요. 돌이켜보면 그게 제일 중요한 거 같아요. 다른 친구들에게도 말하고 싶은 게 목표를 세웠다면 시간이 좀 걸리거나 돌아가더라도 나아가 보라는 거예요. 언젠가는 길이 열릴 거라고 봐요.”
p122-125. 대기업 IT전문가로 조기퇴직 후 10년간 계약직 그러나 IT분야에서 포기하지 않고 곁눈질하지 않고 끝까지 버팀 – 우연한 기회에 후배의 얘기로 스마트공장사업단 전문위원이 됨 – 신영식 (가명) 님 “준비된 자에게는 우연도 기회가 된다.“
p129-133. 자동차회사 영업직에서 사업 (초기 성공 후 영업환경 변화로 실패) 호텔리어로 변신.
p145. 창업시 가장 큰 실수는 동일한 일을 해도 조직의 배경과 체계적 시스템, 동료 협력 등을 바탕으로 일정한 결과를 만들어 내는 것과, 창업을 통해 사업자로서 아무런 배경이나 시스템이 없이 모든 것을 스스로 다 해야 하는 것의 차이를 깨닫지 못하는 것이다.
p148. 창업은 수익성보다는 안전성과 지속성에 집중해야한다. 10-15년간 지속할 수 있는 장수아이템이어야 한다.
안정적 생활을 위한 최소한의 수입을 유지하는 것에 초점을 두고, 해당 수익 목표를 세워서 실천하려고 노력해야 한다.
p150. 본인이 정말 좋아서 하는 일이어야 전문성이 생기고, 이를 지속적으로 발전시켜야 사업의 전문화를 이룰 수 있다.
p174-179. 신창용님의 창업성공사례
팬텍에서 1년간 다시 현역처럼 일에만 몰두하며 신규 프로젝트를 성공적으로 이끌어 낸 그는 회사에 급여와 근무 조건을 조정해 줄 것을 제안했다. 자신이 먼저 급여를 낮추되 일주일에 2~3일만 근무하는 것으로 조정해 달라고 요구한 것이다. 충분한 여유를 두고 퇴직 이후를 준비하고 싶었기 때문이다. 회사에서도 합리적인 제안이라고 판단해서 수락했고, 그때부터 1년간은 일과 퇴직 준비를 병행하며 여유로운 시간을 보낼 수 있었다. “두 번의 퇴직을 통해 많은 걸 얻었어요. 준비 안 된 퇴직의 위험성, 체계적으로 준비하는 퇴직의 안정성 두 가지를 모두 경험할 수 있었던 거죠.”
경험을 통해서 그가 얻은 소중한 삶의 지혜는 늘 미리 ‘다음’을 준비해야 한다는 것이었다. 항상 현재에 안주하기보다는 다음 단계의 커리어를 지속적으로 계획하고, 목표한 바를 이루기 위해서는 조급함을 버리고 충분 한 시간을 들여 준비해야 한다는 것이다.
Viking Therapeutics는 Amgen Scientist였다가 Analyst였던 Brian Lian 박사에 의해 2012년에 설립되었고 Ligand Pharmaceuticals의 5개의 약물 파이프라인을 $2.5 Million Bridge Loan으로 인수함으로써 단숨에 2개의 임상 파이프라인을 포함한 약물군을 확보했습니다.
SAN DIEGO–(BUSINESS WIRE)–Ligand Pharmaceuticals Incorporated (NASDAQ: LGND) announces the licensing of rights to five programs to Viking Therapeutics, Inc., a clinical-stage biopharmaceutical company focused on the development of novel, first-in-class or best-in-class therapies for metabolic and endocrine disorders.
The therapeutic programs covered in the license agreement include Ligand’s FBPase inhibitor program for type 2 diabetes, a Selective Androgen Receptor Modulator (SARM) program for muscle wasting, a Thyroid Hormone Receptor-ß (TRß) Agonist program for dyslipidemia, an Erythropoietin Receptor (EPOR) Agonist program for anemia, and an Enterocyte-Directed Diacylglycerol Acyltransferase-1 (DGAT-1) Inhibitor program for dyslipidemia. The FBPase Inhibitor program was the subject of an option originally granted to Viking in 2012.
Each licensed program includes a fee to be paid to Ligand in Viking equity at the time of a private or public financing, milestone payments and royalties on future net sales. Viking is responsible for all development activities under the license.
As part of this transaction, Ligand has agreed to extend a $2.5 million convertible loan facility to Viking that can be used to pay Viking’s operating and financing-related expenses.
“This is a creative licensing transaction that combines a bold portfolio of early- and mid-stage assets with a company that can advance these programs to major inflection points in the near-term. Viking’s programs have the potential to generate substantial news flow over the next 12 to 24 months and to be the basis for important new drugs in major therapeutic categories,” said John Higgins, President and CEO of Ligand Pharmaceuticals.
“R&D success has been the backbone of our prolific out-licensing activities over the past few years. Our objective is to establish proof-of-concept and solid initial data packages, and then to partner with companies that are well-positioned to manage advanced clinical and regulatory development. A relationship such as this one with Viking gives Ligand the opportunity to entrust valuable internal programs to a dedicated team with the operational resources to take them to the next level. Each of these licensed programs has the hallmark of quality that has defined Ligand’s successful research heritage over the years. We are pleased to have helped establish a platform to advance the programs and to make this investment in Viking to support further progress,” Higgins continued.
“Along with our partners at Ligand, we have created through this license an excellent vehicle to develop several promising new therapies for patients, while unlocking potential value for stakeholders,” said Brian Lian, President and CEO of Viking Therapeutics. “Each of the licensed programs has what we believe to be first-in-class or best-in-class characteristics and a differentiated therapeutic profile. Importantly, the portfolio fits well within Viking’s focus, as our team has an extensive history in diabetes and endocrine drug development, including two recent drug approvals. At all levels, from preclinical through pharmaceutical development, and including our chief medical officer, we have well-aligned development expertise to bring these programs forward.”
About Viking Therapeutics, Inc.
Viking Therapeutics is a clinical-stage biotherapeutics company focused on the development of novel, first-in-class or best-in-class therapies for metabolic and endocrine disorders. Viking’s research and development activities leverage its expertise in metabolism to develop innovative therapeutics that improve patients’ lives. Viking has a portfolio of five drug candidates in clinical trials or preclinical studies, which are based on small molecules licensed from Ligand and its affiliate. Viking’s lead clinical program is VK0612, a first-in-class, orally available drug candidate for type 2 diabetes (Phase 2b). Viking’s second clinical program is VK5211, an orally available, non-steroidal selective androgen receptor modulator, or SARM, for the treatment of cancer cachexia (Phase 2). Viking is also developing three novel preclinical programs targeting metabolic diseases and anemia.
Ligand is a biopharmaceutical company with a business model that is based upon the concept of developing or acquiring royalty revenue generating assets and coupling them to a lean corporate cost structure. Ligand’s goal is to produce a bottom line that supports a sustainably profitable business. By diversifying our portfolio of assets across numerous technology types, therapeutic areas, drug targets and industry partners, we offer investors an opportunity to invest in the increasingly complicated and unpredictable pharmaceutical industry. In comparison to its peers, we believe Ligand has assembled one of the largest and most diversified asset portfolios in the industry with the potential to generate revenue in the future. These therapies address the unmet medical needs of patients for a broad spectrum of diseases including diabetes, hepatitis, muscle wasting, Alzheimer’s disease, dyslipidemia, anemia, asthma and osteoporosis. Ligand’s Captisol platform technology is a patent protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs. Ligand has established multiple alliances with the world’s leading pharmaceutical companies including GlaxoSmithKline, Onyx Pharmaceuticals (a subsidiary of Amgen Inc.), Merck, Pfizer, Baxter International, Eli Lilly & Co. and Spectrum Pharmaceuticals. Please visit http://www.captisol.com for more information on Captisol. For more information on Ligand, please visit http://www.ligand.com.
Ligand Pharmaceuticals has licensed the rights to five programs to Viking Therapeutics, a clinical-stage biopharma company focused on the development of therapies for metabolic and endocrine disorders. As part of this transaction, Ligand will extend a $2.5 million convertible loan to Viking that can be used to pay Viking’s operating and finance expenses. Ligand will receive a fee for each program, milestone payments and royalties on future sales. Viking is responsible for all development activities under the license.
The therapeutic programs include Ligand’s FBPase inhibitor for type 2 diabetes, a Selective Androgen Receptor Modulator (SARM) program for muscle wasting, a Thyroid Hormone Receptor-β (TRβ) Agonist for dyslipidemia, an Erythropoietin Receptor (EPOR) Agonist for anemia, and an Enterocyte-Directed Diacylglycerol Acyltransferase-1 (DGAT-1) Inhibitor program for dyslipidemia.
“This is a creative licensing transaction that combines a bold portfolio of early- and mid-stage assets with a company that can advance these programs to major inflection points in the near-term. Viking’s programs have the potential to generate substantial news flow over the next 12 to 24 months and to be the basis for important new drugs in major therapeutic categories,” said John Higgins, president and chief executive officer of Ligand Pharmaceuticals.
“Along with our partners at Ligand, we have created through this license an excellent vehicle to develop several promising new therapies for patients, while unlocking potential value for stakeholders,” said Brian Lian, president and chief executive officer of Viking. “Each of the licensed programs has what we believe to be first-in-class or best-in-class characteristics and a differentiated therapeutic profile. Importantly, the portfolio fits well within Viking’s focus.”
2015년에 $24 Million IPO를 했습니다. Micro-cap Biotech IPO였지요.
SAN DIEGO–Ligand Pharmaceuticals Incorporated (NASDAQ: LGND) partner Viking Therapeutics, Inc. (NASDAQ: VKTX) announced that it has priced its initial public offering of 3,000,000 shares of its common stock at an initial offering price to the public of $8.00 per share. Viking has granted the underwriters a 30-day option to purchase up to an additional 450,000 shares of common stock at the same price to cover over-allotments, if any. Viking expects the shares to begin trading on the Nasdaq Capital Market on April 29, 2015 under the ticker symbol “VKTX”. Viking expects the offering to close on May 4, 2015, subject to the satisfaction of customary closing conditions.
Laidlaw & Company (UK) Ltd. is acting as the sole book-running manager for the offering. Feltl and Company, Inc. is serving as co-manager for the offering.
Ligand invested $9 million in the offering. Ligand will issue an additional press release once the transaction has closed.
IPO 한 지 근 10년이 지나 Dual GLP-1/GIP Peptide Agonist인 VK2735의 임상2상 결과가 나왔는데 경쟁 약물인 Eli Lilly의 Tirzepatide와 거의 같고 Novo Nordisk의 Semaglutide보다는 나은 체중감소를 보였습니다.
Pass the mead, because Viking Therapeutics has plenty to celebrate after its dual agonist of GLP-1 and GIP was linked to weight loss of up to 14.7% after 13 weeks of treatment, sending the biotech’s stock soaring.
Dose-dependent reductions in the weight of recipients of VK2735, which ranged from 9.1% to 14.7%, were all significantly larger than the 1.7% dip tracked in participants on placebo. While that was enough for the phase 2 study to meet its primary endpoint and clear the 8% bar Viking set as an internal hurdle, the comparison to other molecules is equally important in the increasingly competitive obesity space.
VK2735 looks good compared to the incumbents, with usual caveats about cross-trial comparisons. After 13 weeks, people in Novo Nordisk’s semaglutide phase 3 trial were yet to lose 10% of their body weight. Eli Lilly’s tirzepatide triggered faster, deeper weight loss than semaglutide, but VK2735 looks competitive against that molecule, too. That could catch the eye of pharma dealmakers—and Viking is open to talks.
“Our plan is to proceed aggressively with further clinical development. We’re always open … to [business development] discussions,” Viking CEO Brian Lian, Ph.D., said on a call with investors to discuss the data. “Right now we’re really focused on next steps with the program for us and remain with the ‘open door’ policy for discussing opportunities.”
Would-be buyers may need to dig deep to prise VK2735 away from Viking or buy the biotech outright. The company ended yesterday with a market cap of $3.9 billion but saw its stock climb 80% to above $69 in premarket trading.
Investors sent the stock skyward as they digested data from a 176-subject phase 2 trial that paints Viking as a serious force in the red-hot obesity space. Up to 88% of patients on VK2735 experienced weight loss of 10% or more, compared to 4% of people on placebo, and Viking believes further weight loss is possible beyond Week 13.
“There wasn’t really an indication yet of a plateau signal. The two higher doses, the 10 and the 15, one of them appeared to maybe be accelerating a little bit, but it’s hard to know. These are weekly reads, so you get a little bumpiness as the curve evolves,” Lian said.
Asked by an analyst why VK2735 may outperform the competition, Lian pointed to a pharmacokinetic profile that “provides very good exposures” and a half-life that is “quite long.” Lian said those connected factors may help “drive this level of efficacy.”
The next step is to hold a meeting with the FDA, something Lian expects to happen around the middle of the year. “It seems more than likely that a phase 2b will be the next step here but we’ll have a better idea after we speak with the FDA and get some guidance,” the CEO said.
On the safety and tolerability front, the discontinuation rate across all VK2735 doses was slightly lower than in the placebo group, with 13% playing off against 14%, although the rate at the highest dose was 20%.
As with other GLP-1 drugs, many patients experienced nausea, with the rate peaking at 63% at the top dose, but most of the cases were mild and none were severe. One patient went to hospital with symptoms of dizziness. The patient was diagnosed with dehydration and admitted to the hospital, triggering a serious adverse event, but then recovered.
Viking is yet to share data on markers such as liver fat and plasma lipids, but earlier studies suggest the candidate may benefit patients with comorbidities linked to obesity. Lian said the mechanism “seems to be applicable” to metabolic dysfunction-associated steatohepatitis, an indication Viking knows well from its work on VK2809, but, for now, the biotech is going to direct its resources to obesity.
이 결과를 바탕으로 Public Funding을 했는데 목표했던 $550 Million보다 큰 $632 Million에 마감되었습니다. 아마도 빅파마와 협상이 진행되고 있는 것 같습니다. 어떤 결과가 나올지 궁금합니다. 최근 VK2735 Oral의 임상1상도 진행 중입니다.
Viking Therapeutics, Inc. (“Viking”) (Nasdaq: VKTX), a clinical-stage biopharmaceutical company focused on the development of novel therapies for metabolic and endocrine disorders, today announced the closing of its previously announced underwritten public offering of 7,441,650 shares of its common stock at a price to the public of $85.00 per share, which included the exercise in full by the underwriters of their option to purchase up to 970,650 additional shares of common stock. The gross proceeds to Viking from this offering were approximately $632.5 million, before deducting underwriting discounts and commissions and offering expenses.
Morgan Stanley, Leerink Partners, William Blair, Raymond James, Stifel and Truist Securities acted as joint book-running managers for the offering. Oppenheimer & Co. acted as lead manager for the offering. BTIG, H.C. Wainwright & Co., Maxim Group LLC and Laidlaw & Company (U.K.) Ltd. acted as co-managers for the offering.
Viking currently intends to use the net proceeds from the offering for continued development of its VK2809, VK2735 and VK0214 programs and for general research and development, working capital and general corporate purposes.
현재 Viking Therapeutics의 파이프라인은 아래와 같이 3가지 입니다. Micro-Cap에서 Unicorn이 되기까지 이 여정이 참 신기하기도 합니다.
Idorsia has spun out of Actelion after Johnson & Johnson wrapped up its $30 billion takeover of the Swiss biotech. The new company starts life with $1 billion in cash, multiple clinical-phase drugs and a deal with J&J, strengths that prompted traders to drive up its share price by 30% in its first hours on the Swiss stock exchange.
Shares in Idorsia began trading at CHF 10 ($10) a piece but quickly soared higher. The stock settled around the CHF 13 mark a few hours after the market opened. That jump was foreseeable. Talking to Reuters, an anonymous trader said the initial CHF 10 “reflects the company’s cash and actually is rather cheap.”
Allschwil, Switzerland-based Idorsia’s $1 billion starting cash position is one of several eye-catching characteristics of the biotech. Unlike almost all newly minted biotechs, Idorsia also has multiple drugs in the clinic, one of which could net it a $230 million fee if J&J opts in.
The J&J agreement covers dual endothelin receptor antagonist ACT-132577, one of four phase 2 assets in Idorsia’s pipeline. Actelion posted data from a phase 2 trial of ACT-132577 last month. The readout linked the experimental therapy to statistically significant reductions in mean diastolic and systolic blood pressure.
J&J can pay $230 million and commit to royalties that range from 20% to 35% to opt-in. The 30-day opt-in countdown will start once Idorsia has shared the data with J&J and held an end-of-phase 2 meeting with the FDA. Idorisa plans to run a phase 3 trial in patients with resistant hypertension.
ACT-132577 is one of a clutch of Idorsia assets edging toward phase 3. Idorsia is a phase 2 dose-finding study away from having the data it needs to move cenerimod into a pivotal trial. Insomnia asset ACT-541468 is set to deliver the phase 2 data Idorsia needs to move it into phase 3 later this year. And regulatory talks in preparation for a phase 3 trial of Fabry disease hopeful lucerastat are taking place. Idorsia plans to start a phase 3 trial of lucerastat next year.
Investigators are also testing cerebral vasospasm treatment clazosentan in a phase 2 trial.
Further back in the pipeline, Idorsia is set on four phase 1 assets, although the long-term status of these programs is unclear
“We will be taking decisions on our phase 1 pipeline assets before the end of the year,” Idorsia CEO Jean-Paul Clozel said in a statement.
Clozel is one of several key executives who have moved from Actelion to Idorsia. The CEO is joined at the biotech by Guy Braunstein, who has taken the role of head of global clinical development.
The FDA blessed Idorsia’s Quviviq (daridorexant), a sleeping pill for those with insomnia. Because the FDA has recommended Idorsia to register Quviviq as a controlled substance, the treatment will not reach the market until May, the company said.
After more than two decades of trial and error and experiments with—by their estimate—25,000 compounds, the husband and wife team of executives at Idorsia, Jean-Paul and Martine Clozel, said they have finally reached the finish line with a drug they believe can be a game-changer.
With 25 million Americans affected by insomnia and only about 30% of them diagnosed, a huge untapped market awaits. But Idorsia has a big task ahead to distinguish Quviviq from Merck’s Belsomra, which has been on the market for seven years, uses the same mechanism and has not had the most successful market.
Quviviq is the first approval for Switzerland-based Idorsia, a company the Clozels established in 2017 after they sold Actelion to Johnson & Johnson for $30 billion.
In two phase 3 studies, Quviviq showed significant improvement over placebo in sleep onset, sleep maintenance and total sleep time, while also reducing daytime sleepiness.
Both Quviviq and Merck’s Belsomra are from the dual orexin receptor antagonist (DORA) class. Belsomra has had difficulty catching on, generating sales of $327 million in 2020. Another DORA, Eisai’s Dayvigo, was launched in 2020.
The difference between Quviviq and the other DORAs, according to Idorsia, is that it keeps people energetic and awake throughout the day. Belsomra’s development was hindered by warnings about next-day somnolence and depression, forcing Merck to cut its dosage to suboptimal levels to gain approval.
Finding a treatment that strikes the delicate balance between providing nighttime restfulness and daytime functioning is why it took so long to develop Quviviq, Martine Clozel, executive vice president and chief scientific officer,said in an interview.
“It really took us many years to arrive [at] a compound with the right duration of action,” Clozel explained.
For decades, insomnia has been treated with sedatives, such as Ambien. The generic versions of these treatments still dominate the market, though they can cause memory problems and morning sedation.
Roughly 20 years ago, scientists found it was the orexin system that keeps the brain awake. This discovery kicked off a new way of targeting insomnia. Quviviq and other DORAs block the binding of the wake-promoting neuropeptides orexins, tuning down overactive wakefulness, as opposed to sedating the brain.
At Actelion, the Clozels developed an insomnia treatment, almorexant, but had to bail on it in 2011 because of safety concerns. After licensing the drug to Midnight Pharma, Actelion got to work on another insomnia treatment.
Ten years later, the Clozels believe they’ve finally got it right. “We believe we’ve hit the jackpot with regard to the dose response” Patty Torr, president and GM of Idorsia U.S., said in an interview, also pointing to the fact that Quviviq is not a brain-sedating-type medicine. The drug will be available in 50 mg and 25 mg doses.
Getting patients to believe in this DORA will require a major marketing effort, which the company has already begun with an awareness program, Alliance for Sleep. Another effort underway is the Seize the Night campaign which will specifically touts combination of daytime and nighttime benefits provided by Quviviq.
The unmet need in insomnia is undeniable. Many are diagnosed but do not seek treatment. Additionally, of those who are prescribed medicine, 25% drop out of the market annually, Torr said.
“There’s this churn going on in the marketplace,” she said. And that spells opportunity for Idorsia.
Up to 500 roles at Idorsia are at risk as the Swiss biotech tries to halve its cash burn while it waits for its approved insomnia treatment Quviviq to pay off.
Facing a “challenging financial situation” due to “lower than anticipated product sales and a difficult global financial environment,” the company has decided to sacrifice its R&D work in order to “maximize the time the company has to deliver commercial success.”
Exactly what will happen is still to be decided. Idorsia will now review its development pipeline and jettison all candidates that can’t be “advanced rapidly and with reasonable financial investment.” Work on those unwanted programs will be either paused or prepared for partnership or out-licensing, the biotech explained in an early morning release Friday.
The ultimate goal is to reduce cash burn at the company’s Allschwil, Switzerland, headquarters by 50% by early 2024.
It means a worrying period of limbo for many employees. Up to 500 positions could be set to go as part of the “cost reduction initiative,” mostly located in the R&D team and related roles. The affected employees will be decided as part of a consultation process due to wrap up by the end of the year.
“I continue to believe that Quviviq can be the success we hope for, but unfortunately it will take longer than originally planned,” the biotech’s CEO Jean-Paul Clozel, M.D., said in the release. “Idorsia’s immediate objective is therefore to maximize the time the company has to deliver commercial success with its products. This means making any funds that are raised last as long as possible by significantly reducing our global cash-burn.”
“The cost reduction initiative together with potential collaborations will give the company the time it needs to realize the value we have created,” Clozel added. “I deeply regret having to launch such an initiative, but we simply cannot sustain current investment levels.”
While Idorsia has previously claimed that Quviviq has become the No. 1 branded insomnia treatment since Idorisa launched it onto the U.S. market in May 2022, delays in securing reimbursement meant it only brought in total net sales of 4.3 million Swiss francs ($4.9 million) in the first quarter of the 2023.
In an earnings report in April, Clozel insisted that when it came to the reimbursement delays, “progress has been made and the situation is steadily improving.”
The company ended March with 212 million Swiss francs ($245 million) in cash and equivalents. Since then, Idorsia has secured bridge financing of 75 million Swiss francs ($86 million) as well as sold its Asia-Pacific rights outside of China to Sosei Heptares.
If its commercial rollout has been slow, Idorsia hasn’t had much luck in the clinic in recent years, either. In 2021, its Fabry disease medicine lucerastat flamed out in a phase 3 trial, while cenerimod missed the primary goal in a phase 2b systemic lupus erythematosus study.The following year saw its selective orexin-1 antagonist ACT-539313 fail to hit its goal in a binge eating disorder study, prompting the company to give up on the indication.
It’s made for a tough period for a company that arrived on the biotech scene with a splash in 2017 by way of a spinout from Actelion, which was bought by Johnson & Johnson. Idorsia launched with $1 billion in cash and a handful of clinical-stage drug candidates—plus a potentially lucrative development deal with J&J for the blood pressure medication aprocitentan, which has continued to show potential in a phase 3 trial.
Shares in the company have been on a steady decline in 2023, hovering around the 6.30 Swiss franc mark this morning from a January opening price 15.33 Swiss francs.
올해 3월에 마침내 Aprocitentan (Tryvio, ACT-132577)가 FDA 승인을 받았습니다.
Idorsia Pharmaceuticals on Wednesday announced that the FDA approved its endothelin receptor antagonist aprocitentan for the treatment of hypertension to reduce blood pressure in adults who had not reached adequate control on other drugs.
Aprocitentan, which will now be sold under the brand name Tryvio, is an oral drug taken once-daily alongside other antihypertensive drugs.
Tryvio is the first FDA-approved medication that targets the endothelin pathway for hypertension. Existing treatments act via different mechanisms, including the regulation of salt and water, the reduction of extracellular calcium influx into cells and the inhibition of the renin-angiotensin-aldosterone axis.
Tosh Butt, president and general manager of Idorsia U.S., in a statement said that Tryvio’s approval will “provide physicians and patients with a novel medicine working in a new pathway” to treat uncontrolled hypertension and provide additional blood pressure control. Idorsia is currently preparing its launch strategy for Tryvio and expects to make the drug available to prescribers during the second half of 2024.
Designed to be orally available, Tryvio is a small molecule drug that works by preventing the binding of the endothelin-1 ligand to its corresponding receptors. This mechanism of action dampens signaling cascades that bear many similarities with pathophysiology of hypertension, according to Idorsia’s announcement.
Tryvio comes with a black box warning for embryo-fetal toxicity, flagging the risk of major birth defects when used during pregnancy. Tryvio should not be taken by patients who are pregnant or currently trying to become pregnant.
Idorsia backed Tryvio’s regulatory bid with data from the Phase III PRECISION trial, which enrolled 730 patients who had systolic blood pressure (SBP) of at least 140 mmHg and who were taking at least three antihypertensive drugs. Before treatment, all patients were switched to a standard background regimen consisting of a calcium channel blocker, a diuretic and an angiotensin receptor blocker.
Results showed that both the 12.5-mg and 25-mg dose levels of Tryvio could significantly reduce sitting SBP compared with placebo after four weeks of treatment. Treatment benefits were consistent across several patient subgroups, including according to age, sex, race, diabetes history and body mass index. Tryvio is only approved at its 12.5-mg dose.
Tryvio’s approval comes months after Johnson & Johnson turned its back on the endothelin receptor blocker, electing in September 2023 to return Tryvio’s worldwide rights to Idorsia. The Indianapolis-based pharma bought into the promise of aprocitentan in 2017, paying $230 million for joint development and sole worldwide commercialization rights.
Under the terms of the 2023 handoff, J&J will still be entitled to 30% of any out-licensing proceeds associated with Tryvio, as well as 10% of earnings from any product deal that Idorsia enters following approval.
Lucky genes don’t fully explain super agers’ razor-sharp thinking and memory skills, says Angela Roberts, assistant professor in the School of Communication Sciences and Disorders at Western University in Ontario. “Lifestyle matters,” she says. Here’s what they do, and what you should too:
1. Super agers control their blood sugar and blood pressure.
Super agers tend to have blood pressure and blood sugar levels that are healthier than in the general population. One way to control both is through diet. Older adults who follow an eating pattern rich in vitamin-, carotenoid- and flavonoid-packed foods such as whole grains, veggies, leafy greens, nuts, berries and fish, and low in red meat, butter and sweets slowed brain aging by 7.5 years and kept thinking and memory sharper in a 2015 Rush University study of 960 older adults.
MIND (Mediterranean-DASH Diet Intervention for Neurodegenerative Delay) diet
In a 12-year study published in 2019, this eating strategy lowered the risk for Alzheimer’s disease and dementia by up to 53 percent. If you have high blood pressure or diabetes, talk with your doctor about medications and other strategies to keep these conditions under control. Setting a goal of a systolic bp (the top number in a bp reading) below 120 lowered risk for mild cognitive impairment by 19 percent in one study of 9,361 older adults who took medications for their high blood pressure. For those not taking medication for high blood pressure, it also lowered the risk for either mild cognitive impairment or dementia by 15 percent.
2. Super agers don’t exercise more, but they push themselves physically.
Spanish researchers followed a cohort of 119 people, ages 70–85, for eight years; among that group were 55 super agers who scored at least 20 years younger than their years on brain tests. Researchers found that what distinguished super agers most profoundly was that they have greater speed, mobility, agility and balance than typical older adults — despite reporting the same exercise frequency.
One reason may be that super agers tend to do more demanding and rigorous activities such as gardening or stair-climbing, even though they report similar activity levels to other adults. In other words, walking a mile is good for your health; walking fast for a mile to get your heart rate up is even better. One British study found that just nine minutes of moderate-intensity exercise daily improved thinking skills.
3. Super agers avoid stress and prioritize mental health.
Another top distinguishing factor among super agers in that recent Spanish study: They reported lower levels of anxiety and depression than normal agers.
That makes sense: A recent three-year Danish study found that depression doubled risk for dementia, and a 2023 study found that those with the perceived high stress levels had a 37 percent higher risk for memory problems compared with those reporting low stress levels. Another study found that older adults with depression who got treatment — including medication and talk therapy — were up to 32 percent less likely to develop dementia over 10 to 14 years than those who didn’t get help. A fourth study found that those whose anxiety improved with talk therapy lowered their risk for later dementia by 17 percent.
4. Super agers protect their vision and hearing.
Researchers speculate that the brain may neglect memory processing as it instead puts extra effort into decoding blurry, muted signals from the world around us. A 2022 University of Toronto study of 5.4 million older Americans, age 65-plus, found serious cognitive problems for 28 percent of people with vision loss, 20 percent of those with hearing loss and 50 percent of those who had both poor vision and poor hearing.
Caring for your eyes and ears can pay off: University of Washington researchers found that at-risk older adults who received hearing aids showed thinking and memory losses that were 48 percent slower compared with those who didn’t get hearing aids. Similarly, a study of older adults with cataracts found that those who had cataract surgery had a 29 percent lower risk for dementia for up to 24 years compared with those who did not have the procedure. Getting help for poor vision — such as eyeglasses and cataract surgery — could have prevented 100,000 current cases of dementia in the U.S., according to a 2021 study.
5. Super agers prioritize sleep.
During slumber, your brain clears away toxic waste that builds up early in the development of Alzheimer’s disease. A 2022 Canadian study found that trouble falling or staying asleep three or more nights per week for three months boosted the risk for worsening memory in older adults.
“Good sleep is really important for maintaining brain health,” says Jeff D. Williamson, professor, gerontology and geriatric medicine, Wake Forest School of Medicine, who suggests discussing sleep issues with your doctor. Don’t rely on over-the-counter and prescription sleep drugs on a regular basis. Chronic use of prescription sleep drugs boosted the risk for dementia by 48 percent over six and a half years in a 2021 University of Minnesota study of 4,197 at-risk people in their 70s.
6. Super agers do more than Wordle.
Super agers did crossword puzzles and Sudoku games more often than “normal agers” in the Spanish study. They were also more likely to frequently read, listen to music, go to concerts and movies, travel, play cards and board games, do something creative such as handicrafts or performing in a play, and attend lectures. “Variety is beneficial,” says brain-game researcher Aaron Seitz, professor of psychology, physical therapy, art and design at Northeastern University. “Your brain needs to do a lot of different things. If we want to do them well, science and common sense suggest exercising it in a lot of different ways.” Super agers tend to move out of their comfort zones and share a willingness to endure discomfort to master a new skill such as playing a musical instrument or learning a language.
7. Super agers talk to their friends — a lot.
Older adults who connected every day with others had less shrinkage in key brain areas than those who seldom had contact with pals and relatives, according to a 2023 Japanese study in the journal Neurology. Perhaps that’s why memory declined fastest and furthest in people who felt lonely most often, in a 2022 University of Michigan study that tracked 9,032 U.S. adults for 20 years.
신중년 개개인들은 어느 날 자신이 부모세대와 더 오래 살게 되면서 다른 삶을 살게 된다는 점을 부지불식간에 인식한다. 그런 신중년들을 만나보면 대부분 정년 이후의 삶, 특히 일에 대한 걱정을 많이 하고 있다는 특징이 발견된다. 그런 걱정은 대부분 일자리 부족, 나이에 대한 사회적 차별, 개인의 자존감 및 건강상태 등 다양한 사회적 혹은 개인적 문제로부터 출발한다.
우리 사회에서는 오래전부터 ‘100세 시대’라는 이야기가 들려왔고, 최근에는 ‘120세 시대’라는 이야기도 심심치 않게 들리고 있다. 이에 국가에서도 이전과 달리 인생 전반부를 마치는 신중년들의 후반부 삶을 지원하기 위해 ‘1,000인 이상의 기업 퇴직자’, ‘50세 이상’ 등을 대상으로 ‘재취업지원서비스 의무화’와 ‘생애(경력)설계’ 실행을 위한 정책을 펴고 있다. 따라서 미래에는 어떠한 형태로든 더 오래 일하게 될 신중년들에게 도움을 주고자 근로 생애에 대한 새로운 관점 3가지를 제시해본다.
첫 번째 관점 : 일자리가 아닌 일거리의 시대가 온다. 인생 후반에 들어선 신중년 대다수는 가능하다면 계속해서 일하고자 한다. 부모세대와 달리 의약품, 과학기술 및 섭생의 개선으로 더욱 건강해지고 수명도 연장됐기 때문이다. 그런데 막상 인생 후반부에 들어서면 인생 전반부와 같은 온전한 일자리를 찾기 힘들다. 이때는 먼저 일자리를 ‘온전한 직업’으로, 일거리를 ‘삶과 일을 잘 조화시키는 활동’ 개념으로 이해해야만 한다. 일자리의 개념이 ‘완벽한 직업의 개념’이라면, 일거리는 그에 미치지는 못하지만 여전히 ‘일을 하면서 활동하는 개념’이다.
이전 산업화시대의 ‘소품종 대량생산’의 개념이 4차산업혁명시대로 진입함에 따라 ‘다품종 소량생산’의 개념으로 전환되면서 공장생산보다는 개인이 직접 생산하면서 자급자족하는 일거리의 시대로 바뀌고 있다. ‘9시에서 6시’까지 일하는 고착된 일자리의 개념에서 벗어나서 개인이 자신의 근로시간을 다소 자유롭게 선택하는 일거리의 시대가 우리 앞에 성큼 다가왔다. 더불어 하나의 일자리에서 주로 일한 이전의 삶과 달리, 이후의 삶은 개인에 따라 다르겠지만, 하나 혹은 그 이상의 일거리를 가지고 살아가는 개념이다. 박영숙 유엔미래포럼 한국대표는 “2030년에는 일자리의 시대에서 일거리의 시대로 진입한다”고 강조한다. 삶과 일이 더욱 자유로운 일거리의 시대에 대비해보자!
두 번째 관점 : 100세 시대 근로생애는 80세까지다. 현재의 신중년들은 인생 후반을 맞이하면서 전환과 관련된 각종 학습을 하는데, 과연 그들의 부모세대도 그러했을까? 아니다. 신중년들은 자신의 부모보다 더 긴 시간을 살면서, 더 긴 근로생애를 맞이하기 때문에 그에 대비한 평생학습을 해야만 한다. 예를 들어 평생학습은 이전의 힘든 학교공부가 아니라 ‘핸드폰 잘 사용하는 법’ 등과 같은 세상의 변화를 따라가는 학습으로 보면 좋다.
그렇다면 언제까지 일해야 할까? 많은 신중년이 인생 후반에 접어들면서 갖게 되는 의문이다. 현장에서 강의나 컨설팅을 하면서 신중년들에게 질문해보면 대부분 70세 정도까지 일할 것이라고 답변한다. 그러나 앞서간 신중년 선배들은 “상황이 허락하는 한 일을 계속하겠다”고 각종 설문 조사에서 밝히고 있다. 영국 런던대학교의 린다 그래턴 박사는 그의 저서 ‘일의 미래’에서 다음과 같이 이야기하고 있다. “이전에는 80세까지 살고 60세까지 일했다면, 이제는 100세까지 살고 80세까지 일해야 한다. 그를 위해서는 지속적인 학습이 필요하다”고 주장한다. 100세를 넘어선 철학자 김형석 교수도 그의 저서 ‘백년을 살아보니’에서 “80세까지 일하는 사람은 매우 행복한 사람이다”라고 밝히고 있다. 자신의 근로 생애를 스스로 80세까지 멀리 잡고 그에 대한 준비를 하자!
세 번째 관점 : 80세 근로생애의 중간에 일의 변곡점이 있다. 근로생애가 이전의 부모세대보다 연장된다고 하니 힘들게 인생 전반을 보낸 신중년들은 “나보고 또 힘들게 일하라고?” 혹은 “그렇게 오랫동안 일해야 하나?” 또는 “계속해서 일하기 위해서 또 공부해야 한다고?”라고 볼멘 목소리를 낸다. 그러나 80세 근로생애는 일하는 여정에서 일의 변곡점을 만나는데, ‘힘든 일자리’에서 ‘여유로운 혹은 자유로운 자신만의 일거리’의 개념으로 넘어가는 점이다.
이는 각자의 가족부양이나 자녀교육을 마친 이후로 부담 없이 자신이 원하는 곳에서, 원하는 일을 하면서 진정한 자아실현을 하는 개념으로 보면 된다. 개인의 상황에 따라서 차이는 있겠지만, 대부분 50세 이후에 부담 없이 일할 수 있는 변곡점이 나타나기 시작한다. 예를 들어 자녀에 대한 의무 이행으로 볼 수 있는 대학입학, 대학졸업, 취업, 결혼 등의 시점까지를 자신이 열심히 일하는 기간으로 보고, 그 변곡점 이후는 일자리 혹은 일거리에서 정말 자신을 위해 일한다는 개념이다. 이를 위해서는 먼저 부부간 대화를 통해서 여러 가지 상황을 아우르는 변곡점을 설정하고, 이후 자녀들과의 유연한 대화를 통해 그런 상황을 설명하는 것이 좋다. 그 변곡점이 올 때까지 열심히 일하고, 그 이후는 자신이 진정 바라던 일을 해보자!
이전과 달리 오래 살게 된다면 그에 따라 개인적인 삶의 패턴도 변화돼야만 한다. 힘들다기보다는 오히려 자신의 꿈을 인생 후반에서 펼칠 수 있는 기회가 될 수 있다는 긍정적인 마음을 가지고 길어진 근로생애를 맞이해보자. 자신을 위한 일자리 혹은 일거리가 기다리고 있기 때문에 인생 전반부에서 경험하지 못한 행복한 인생의 황금기를 맞이하게 된다. 앞서 언급한 김형석 교수님도 “60세에서 75세까지가 인생의 황금기”라고 말했다. 신중년을 기다리고 있는 그 황금기를 만끽해보자.
신중년 대부분은 인생 1막에서 하나의 온전한 일자리를 가지고 살아오다가 인생 2모작 혹은 인생 다모작의 삶으로 진입한다. ‘새 술은 새 부대에’라는 말이 있다. 이를 인용해서 비유해보면 ‘새 부대’는 새로운 사고방식이나 탐색방식이고, ‘새 술’은 새로운 일자리, 일거리로 이야기할 수 있다.
인생의 전환기를 맞는 사람이라면 누구라도 ‘새 부대’(=생각의 전환)에 ‘새 술’(=다양한 일자리, 일거리)을 담아보려는 희망적인 생각을 해야만 하지 않을까. 그러나 고용시장에서 보는 다수의 신중년은 인생 1막의 고정관념, 즉 재취업이나 창업 혹은 ‘하나의 온전한 일자리’를 지향하는 경우가 있다. 그런 생각을 하는 신중년들의 건강한 이후 삶을 위해서 ‘생각 열기’를 권하고 싶다.
생각 열기, 생각 확장하기
첫째, ‘새 부대’는 ‘시대의 변화에 따른 일자리, 일거리 개념의 재정립’이다. 인생 1막에서는 ‘하나의 일자리’에서 근무했다면, 그 이후에는 ‘여러 개의 일거리’ 개념으로 출발할 필요가 있다. 더 이상 다수가 정규직으로 일할 수 있는 세상이 아닌 계약직, 파트타임, 시간제, 프리랜스 등의 이름으로 일하는 세상이 왔기 때문이다. 인생 1막의 온전한 ‘하나의 일자리’가 ‘1’이라는 수입이나 일하는 시간적 볼륨을 안겨줬다면, 이후의 삶은 ‘여러 개의 일자리’에서 ‘0.1, 0.3….’ 등의 수입을 만들어가면서 온전한 ‘1’을 지향해 나가야만 한다. 좀 더 확장해보면 이전에는 생계 목적, 가족부양, 부모님의 결정 등 때문에 ‘1’을 지향하면서 일했다면 이제는 세상의 변화를 수용하는 자신의 결정에 따라 ‘0.1….’에서부터 출발한다는 개념이다. 물론 개인에 따라 출발점은 다를 수 있다.
둘째, ‘새 술’은 ‘다양하게 일하는 방식’인데, 첫째 항의 일자리, 일거리 개념을 적용하는 방식이다. 이는 인생 1막에서 주로 고려하던 취업, 창업 그리고 조금 확장된 귀농을 벗어나는 개념이다. 세상이 다양화되는 만큼 일하는 방식도 아래 그림처럼 다양해졌다는 사실을 인식하고, 다양함을 추구해야만 한다. 이전과 달리 지역에서 생산되는 ‘막걸리’만 마시는 것이 아니라 전국의 모든 막걸리를 어느 지역에 살더라도 쉽게 만나고, 쉽게 마실 수 있는 것과 같은 개념이다.
재취업의 경우에는 통상적인 동종산업?동일직무로 전환하는 것뿐만 아니라 동종산업·다른직무, 다른산업·동일직무, 다른산업·다른직무도 고려해보는 것이다. 1인 기업은 쉽게 이야기해서 자신의 서비스나 생산품을 판매하는 프리랜스를 생각해보면 되는데, 자신의 전문성에 기초하여 혼자 혹은 소수와 같이 보유한 지식 등에 기초하여 일하는 방식이다.
전문가 창업은 몇 명의 전문가가 보유한 동종 혹은 이종의 전문성에 기초해 협업기업을 구성하는 형태이다. 요즘 사회적 기업, 협동조합이 그런 협업기업의 주류를 이루고 있다.
전문계약직은 자신이 지닌 전문성에 기초해 특정 프로젝트를 일정 기간 계약 하에 수행하는 형태인데, 고용주의 고용부담도 덜면서 전문가들의 전문성을 활용하는 개념이다.
창직은 4가지로 분류하는데, 창조하거나 새로이 발견, 기존의 직업을 분화시키거나 2개 이상의 기존직업을 융합해 특정한 솔루션을 가지고 새로운 직업을 창출하는 것이다. 사회공헌의 경우에는 자신의 전문성을 사회에 환원, 보답하는 형태인데, 새로운 일자리, 주로 일거리로 옮겨갈 수 있는 기초 역할을 해주는 징검다리 역할을 한다.
귀농, 귀산, 귀어 등은 새로운 장소에서 새로운 삶을 구사하는 것으로서 유의할 점은 반드시 ‘아이템’과 ‘판로’를 어느 정도 가늠한 이후에 실행해야만 한다. 따라서 창업의 개념으로 바라보아야만 한다.
창업의 경우도 통상적인 창업을 벗어나서 기술창업, 프랜차이즈 창업, 외주창업 등으로 구분한다. 마지막으로 제3섹터는 공공기관이나 민간영리부분에서 수행하기 힘든 틈새의 일을 노리는 형태로서 주로 사회적기업, 협동조합을 만들어서 비영리업무를 수행하는 형태이다.
셋째, ‘새 술’을 담은 ‘새 부대’를 끌고 나아가는 방법인데, 이전과 달리 이제는 자기 자신을 ‘경력사업가’로 생각해야만 한다. 대부분 인생 1막에서는 내가 하고 싶은 일을 한 것이 아니라 남이 주는 일을 한 경우가 많다. 이제 인생 2막부터는 자신이 자신을 고용하는 형태로 시작해보자. 따라서 이제는 자신의 경력을 직접 구상하고 운영하는 ‘경력사업가’ 마인드를 가지고 출발해야만 한다. 다시 말하면 사업가적 마인드로 자기 경력을 만들고 거기에 시간, 노력 그리고 금전을 투자하면서 궁극적으로 경력 가치를 향상시키는 형태다.
이제는 이전과 달리 온전한 한 가지가 아닌 여러 가지의 일을 다양한 방식으로, 자기 주도적으로 꾸려나갈 시간이 왔다. 필자는 고용시장에서 꾸준히 자신이 판매할 수 있는 ‘서비스’나 ‘상품’을 개발하고, 이를 ‘기업’, ‘단체’ 그리고 ‘개인’에게 판매하면서 자신의 삶을 개척해나가는 신중년을 종종 만난다. 최초에는 여러 가지 실패를 맛보는 때도 있지만, 어느 정도 시간이 지나면 온전한 ‘경력사업가’로 거듭나는 경우를 종종 본다. 머지않아 그들에게 남의 세상이 아닌 자신의 세상이 도래할 것이다.
다음은 한창묵 (성균관대학교 행정학과 박사과정) 님이 2019년에 발표하신 논문 “신중년의 성공적 노화에 미치는 영향요인 분석”입니다.
저출산 고령화로 인해 장기적 경제성장률이 낮아질 것으로 예상되는 가운데 OECD 국가를 비롯한 각국에서는 이민정책이나 탄력근로제, 워라벨 개선 등과 같은 방식으로 젊은노동력을 확보하고자 노력하고 있습니다. 하지만 이것은 시간이 걸리는 문제이고 중장기적으로는 정년연장이나 계속고용과 같은 방식으로 50대 이후 근로자들을 오랜 기간 직장에서 근무하게 하는 것이 대안으로 거론됩니다. 이런 것은 초고령화가 가장 빠른 일본에서 지속적으로 정년연장, 정년폐지 혹은 계속고용과 같은 방식으로 일하는 연령을 기존 60세에서 차츰 늘려서 이제는 70세까지 일하도록 권고하는 방식으로 나아가는 것을 볼 때 다른 나라들도 이와 유사한 경로를 따를 것으로 생각합니다.
결국 중요한 것은 50대 이후 근로자들의 생산성을 향상시킬 수 있느냐에 대한 방안들이 될 것 같아서 이에 관한 논문을 가지고 그 결과를 좀 나누고자 합니다.
첫번째 논문은 Alliance for Organizational Psychology 에서 나온 자료로 제목은 “Active Aging at Work (직장에서 활동적 노화)” 입니다.
Active Aging (활동적 노화) 의 정의는 WHO에서 다음과 같이 정의했습니다.
The process of optimizing opportunities for health, participation, and security in order to enhance quality of life as people age. (나이듦에 따른 삶의 질을 개선하기 위해 건강, 참여, 보장에 대한 기회들을 도모하는 과정)
Active Aging을 위해
Maintain or improve their physical, mental, and social well-being (육체적, 정신적, 사회적 활력을 유지하거나 개선해야 하고)
Continue to show high levels of work engagement and performance (지속적으로 직장에서 적극적으로 참여하고 높은 업무능력을 보여주며)
Experience fair treatment and employment security. (공정한 대우와 고용 안정을 이루는 것입니다)
활동적 노화 (Active Aging)에는 4가지 요인이 있다.
개인적 요인 (Individual factors): 육체적/정신적 능력, 개인 특성, 신념/신앙, 나이에 따른 삶의 목적 변화
업무/팀 요인 (Job and Team Factors): 축적된 업무지식 및 경험을 활용할 때 실적이 향상, 일의 의미부여, 다양한 연령대의 팀에서 일할 때 만족감을 느낌
기업조직 요인 (Organizational Factors):
Depreciation model: 직원은 특정 연령에서 정점을 이루고 나이가 듦에 따라 성과가 감소한다는 모델 – 조기퇴직을 유도하는 인사정책을 펼
Conservation model: 적절하게 교육받고 관리하면 직원은 지속적으로 성과를 낸다는 모델
비업무적 사회적 요인 (Nonwork and Societal Factors): 가족돌봄이나 사회공헌 등으로 인한 업무 성과 영향 불확실, 나이와 관련한 사회적 통념 및 은퇴 종용 문화로 인한 부정적 요인, Bridge Employment (현직 정년 은퇴 후 계약직 재고용) and Self-Employment (ex. Consulting) 가 주는 긍정적 요인
직장내 활동적 노화를 위해 기업이 할 일
나이든 직원 채용 및 유지 (Recruiting and Retaining Older Workers)
경력관리: 업무영역을 확장하거나 본인에게 흥미로운 일에서 성과를 냄
교육개발: 자신의 교육 투자에 대한 자치권 중요, Self-paced training/self-directed informal learning is a major factor in an organization’s success.
Work Design and Health and Performance Management: Older workers value autonomy, meaningful work, and work allowing them to use their knowledge and skills. Job Crafting (Bottom-up job tailoring for older workers)
Managing the Transitions to Retirement and Bridge Employment: phased retirement, financial retirement planning, succession planning and knowledge transfer.
 나이듦의 8가지 모습")

 Mirati/BMS – Krazati (Adagrasib, KRAS G12C Inhibitor)")


 Themis Bioscience GmbH: Merck’s wholly-owned subsidiary – Recombinant Measles Vector Vaccine Platform")

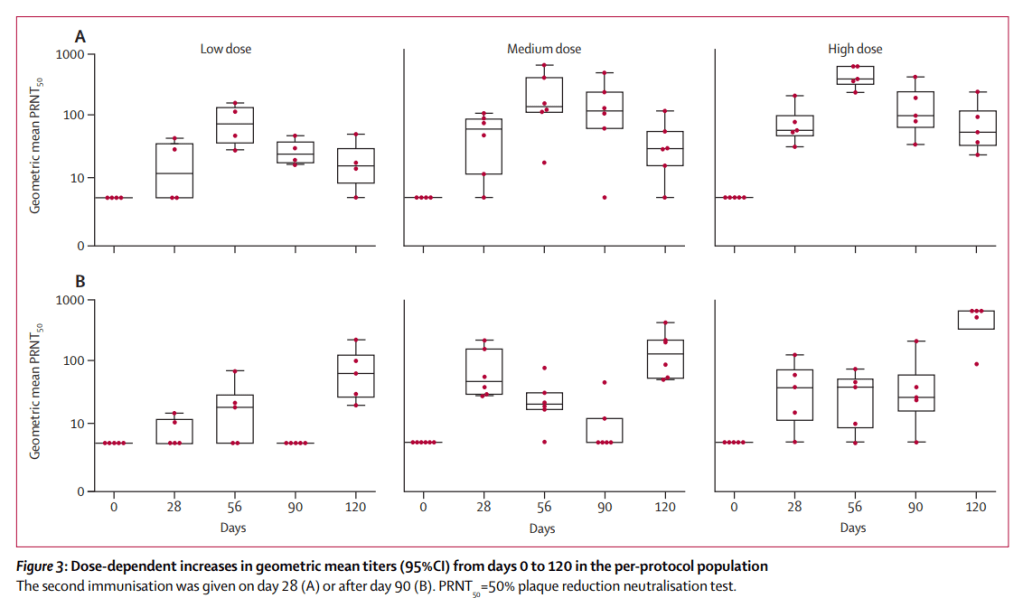
 Omega Therapeutics: Insulated Genomic Domain (IGD) Platform – Controllable Epigenomic Programming Platform")



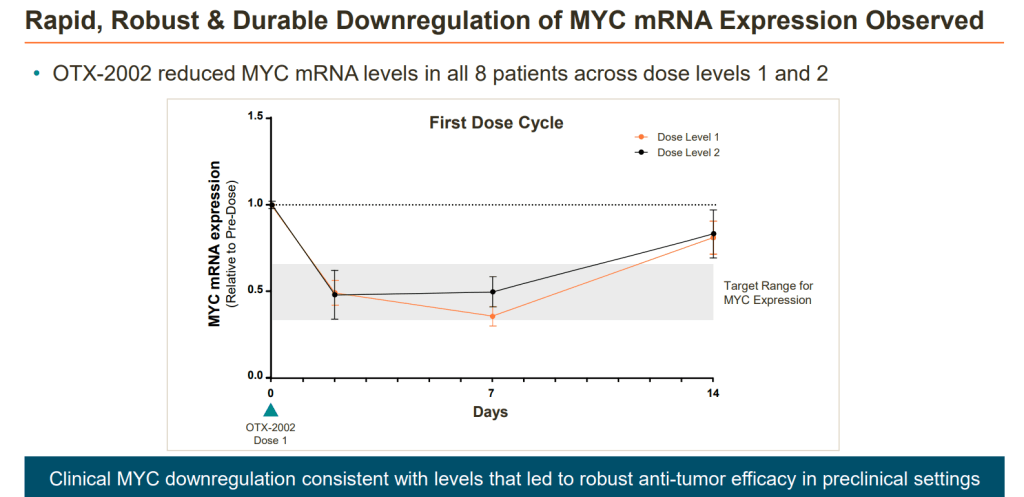

 신중년 역량강화 컨설팅 자료")


 Viking Therapeutics: VK2735 – Dual GLP-1/GIP Agonist")

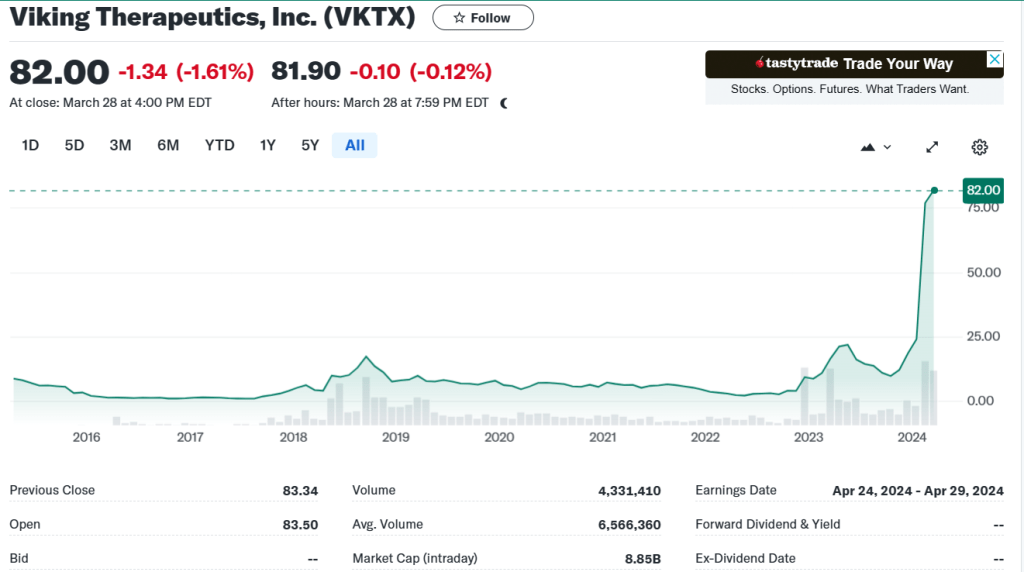
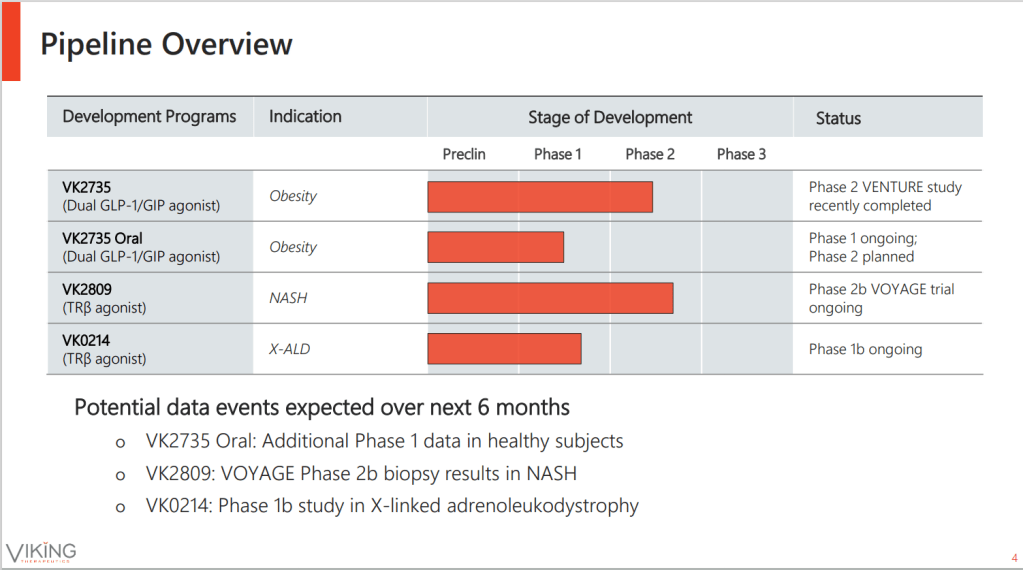
 Idorsia Pharmaceuticals: Johnson & Johnson/Actelion Merger Spinoff – first Endothelin Receptor Antagonist Aprocitentan FDA Approval")

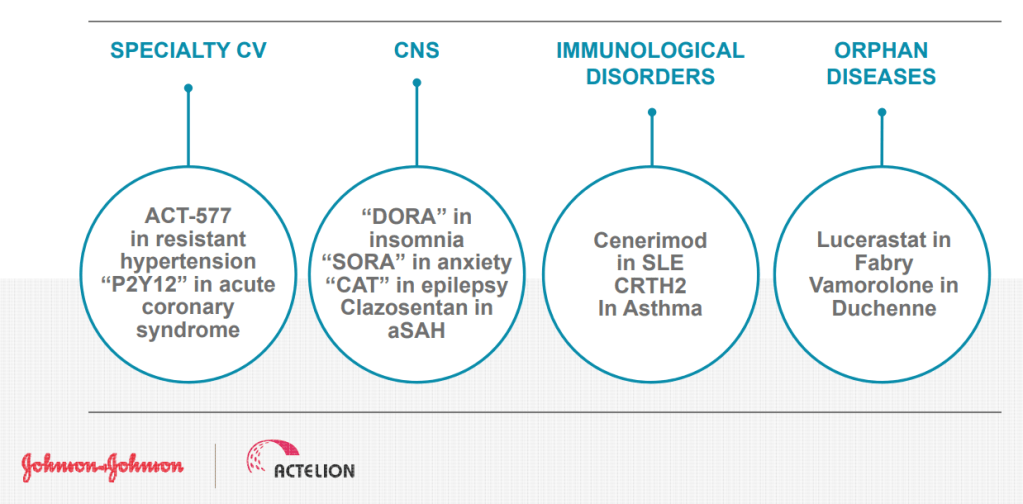
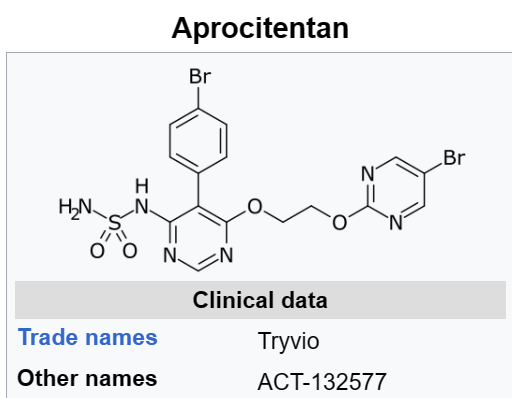
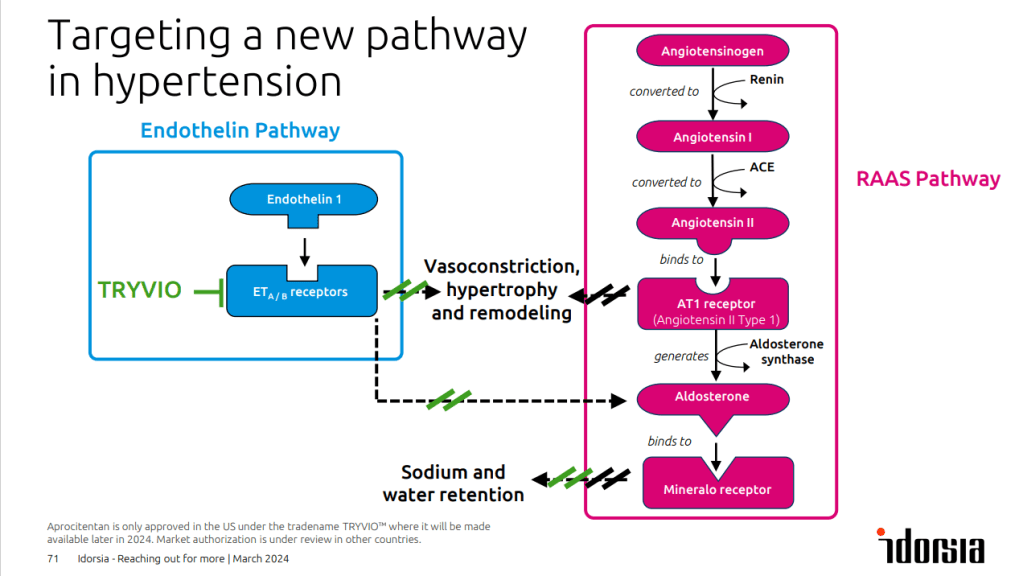
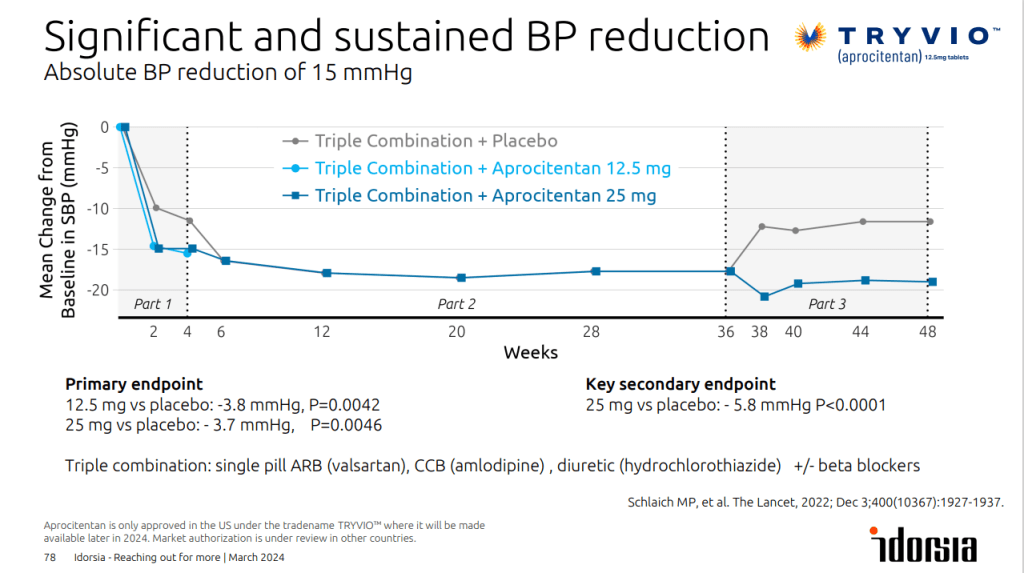
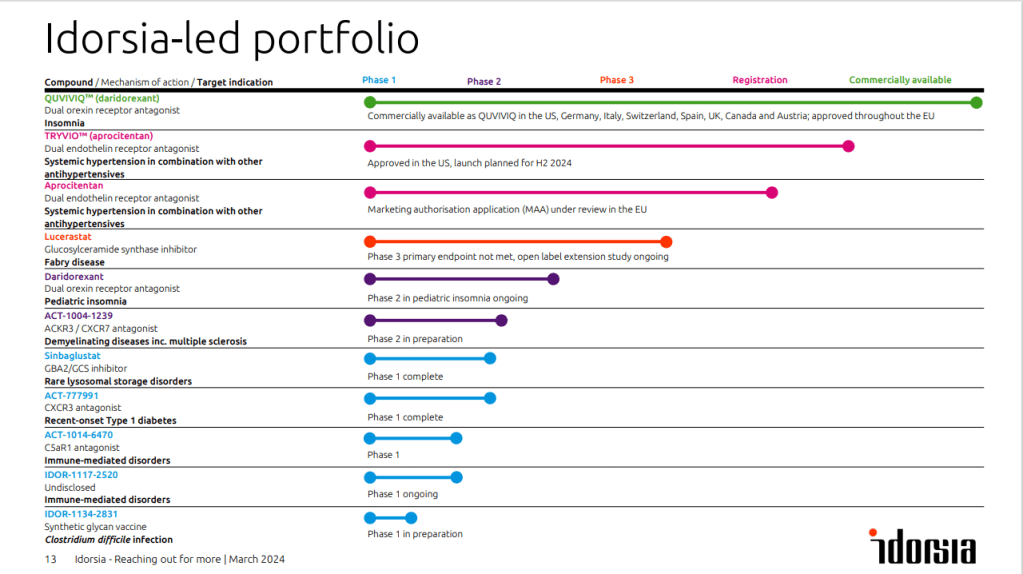
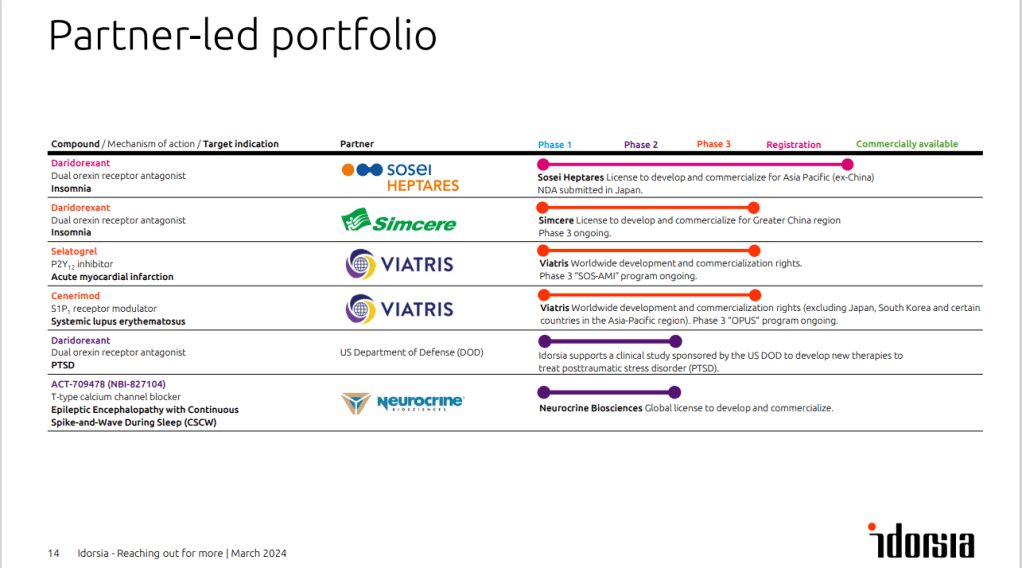
 Super Agers Study – 신경과 정지향 교수 강의 & AARP 7 Secrets")





 신중년의 삶")

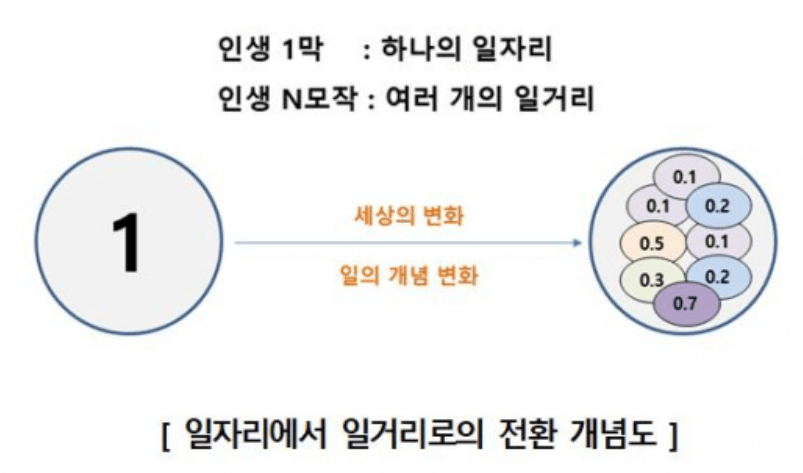


 Active Aging at Work (직장내 활동적 노화)")

