당시에 MDS (myelodysplastic syndrome)에 대한 임상3상 결과가 좋아서 FDA 승인이 기대가 되는 상황이었습니다. 오늘 FDA의 Advisory Committee Meeting이 있었는데 12:2로 승인을 하는 쪽으로 결론이 났습니다. 물론 FDA가 이 결정에 따룰 이유는 없지만 그래도 최종 결정에 긍정적인 결과를 주리라고 기대합니다.’
현재 MDS의 치료제로는 erythropoiesis-stimulating agents (ESA)가 거의 유일한 치료제라고 해도 무방한데 Geron은 Imeltestat을 ESA에 듣지 않는 환자들에 대한 치료제로 승인 요청을 할 예정입니다. Imetelstat에 대한 PDUFA date는 6월 16일입니다.
Members of the FDA’s Oncologic Drugs Advisory Committee voted 12 to 2 on Thursday that the benefits of Geron’s imetelstat outweigh safety risks for the treatment of certain anemic myelodysplastic syndrome (MDS) patients who are dependent on blood transfusions.
While regulators raised concerns around cases of cytopenia, or low levels of white blood cells or platelets, advisory committee members said they were confident that the risks appear manageable. The FDA noted on Thursday that there was a “notably higher” incidence of neutropenia and thrombocytopenia in the imetelstat arm of a Phase III trial.
“Though I am concerned about the risks in this total trial population — in other words, not just the responders — I do believe it is more likely than not that there is a quality-of-life benefit here that is real,” University of Colorado associate professor Christopher Lieu said.
Stanford University School of Medicine professor Ranjana Advani added, “The community of doctors who take care of these patients know how to manage these side effects.”
Members also pointed to the fact that imetelstat met its primary endpoint in a Phase III trial, helping patients achieve eight-week red blood cell-transfusion independence, as well as a key secondary endpoint measuring 24-week independence.
MDS occurs when the normal production of blood cells is disrupted. Most patients with lower-risk MDS experience anemia, which can cause symptoms ranging from fatigue to irregular heartbeat and has also been linked to shorter survival. Those with severe anemia may be dependent on continuous transfusions. Patients who spoke during the adcomm stressed that frequent transfusions impacts their quality of life.
Ravi Madan, a senior clinician head at the National Cancer Institute’s Center for Cancer Research, was one of two members who voted against imetelstat’s benefit-risk analysis.
“I interpreted the question pretty strictly,” he said. “Even low-risk MDS patients are at high risk from their disease, but they shouldn’t also be at risk from their treatments as well.”
The FDA also raised concerns in briefing documents published in advance of the meeting that a majority of patients in the only randomized efficacy trial for imetelstat were enrolled outside of the US. Geron’s chief medical officer Faye Feller acknowledged that a majority of patients were from the EU, but assured the committee that “overall, the demographics are representative of the US MDS population.”
Traditionally, a class of drugs called erythropoiesis-stimulating agents (ESA) have been used off-label to treat lower-risk MDS patients with transfusion-dependent anemia. Bristol Myers Squibb’s Revlimid and Reblozyl are approved to treat anemia in MDS patients, and last year Reblozyl won a label expansion for first-line lower-risk patients who may require transfusions.
But Vanderbilt University School of Medicine professor Michael Savona, who was a member of the steering committee for imetelstat’s Phase III trial, said the use of those drugs is restricted to specific subgroups of patients.
“After failure of ESAs there is no good therapy for most patients,” he said during the meeting.
Geron is seeking approval for patients who have failed on or are ineligible for ESA treatment.
Johnson & Johnson saw early potential in imetelstat, shelling out $35 million upfront and promising another $900 million in milestones to partner on the drug a decade ago. The company backed out of the collaboration four years later, citing “a strategic portfolio evaluation and prioritization of assets.”
GRO Biosciences Inc. today announced that the company has secured $2.1 million in a seed funding round led by Digitalis Ventures and joined by Eric Schmidt’s Innovation Endeavors. The funds will support buildout of bioprocess development for GRO Biosciences’ platform of genomically recoded bacteria for the production of therapeutic proteins with enhanced properties, such as increased potency and stability, and improved targeting and delivery into cells and tissues.
GRO Biosciences was co-founded by the following:
George M. Church, Ph.D., professor of genetics, Harvard Medical School, will serve as head of the company’s scientific advisory board.
Andrew D. Ellington, Ph.D., professor of biochemistry, University of Texas at Austin, will serve on the company’s scientific advisory board.
Daniel J. Mandell, Ph.D., will serve as the company’s CEO.
Christopher J. Gregg, Ph.D., will serve as the company’s chief scientific officer.
P. Benjamin Stranges, Ph.D., will serve as the company’s principal scientist.
Marc J. Lajoie, Ph.D., and Ross Thyer, Ph.D., will serve the company in advisory roles on a consulting basis.
By recoding the genomes of bacterial strains used in biologics production, GRO Biosciences can expand beyond the 20 amino acid building blocks typically found in proteins to introduce non-standard amino acids that can customize the shape and chemical properties of the protein.
“For decades, bacteria have been used as the workhorses of the biotech industry in the production of blockbuster therapeutics, and we believe that we can dramatically expand their utility by recoding their genomes,” said Dr. Church. “GRO Biosciences’ technology addresses the fundamental limitations of producing proteins with non-standard amino acids, opening up the possibility of creating a new universe of designer proteins with enhanced therapeutic properties at commercial scale.”
Nearly all monoclonal antibodies, as well as many other therapeutic proteins, such as interferon, human growth hormone and insulin, used to treat common chronic conditions, rely on disulfide bonds to maintain their 3-dimensional structure needed for biological activity. However, disulfide bonds are not stable in the presence of reducing agents found in cells and in blood, which means that the therapeutic effect of the proteins is short lived after administration to the patient.
(Picture: Andrew D. Ellington at University of Texas at Austin)
GRO Biosciences is addressing the challenge of therapeutic protein instability by replacing disulfide-bond-forming cysteine amino acid residues with selenocysteine, a naturally-occurring amino acid that is rarely incorporated into proteins, but is found in the cell. Selenocysteine has a structure and chemical properties very similar to cysteine, however diselenide bonds are stable under the same conditions where disulfide bonds are not, leading to a much longer half-life of the therapeutic protein.
“Protein therapeutics represent a $180 billion market, yet product stability, targeting and delivery into the cell still remain significant challenges to be addressed if we are to enhance the patient experience, achieve better compliance and improve health outcomes,” said Geoffrey W. Smith, founder and managing partner of Digitalis Ventures. “If GRO Biosciences can make a therapeutic protein product that is more stable and requires less frequent dosing, then that is a win for patients and the healthcare system.”
GRO Biosciences is taking advantage of redundancies in the genetic code to reassign redundant codons to new, non-standard amino acids. For example, there are three different stop codons which are responsible for halting the elongation of a growing protein: UAG, UAA and UGA. GRO Biosciences has developed a recoded strain of bacteria that has replaced all of the UAG stop codons with UAA stop codons and reprogrammed the UAG codon to new amino acids such as selenocysteine. By replacing all codons that code for cysteine residues in a protein with UAG, selenocysteine can be selectively incorporated in place of cysteine residues to form stabilizing diselenide bonds.
“GRO Biosciences is literally reprogramming biology,” said Dror Berman, Managing Partner of Innovation Endeavors. “What I find most compelling is their ability to converge game-changing synthetic biology with powerful computational design to create a new class of living organisms, unlocking unprecedented capabilities in medicine, materials and biotechnology.”
GRO Biosciences has established preliminary proof of concept of its platform by producing diselenide stabilized antibody products as well as therapeutic proteins, such as human growth hormone, in its selenocysteine recoded bacteria. In all instances, yields were high, selenocysteine incorporation at the desired sites was 100 percent, and all diselenide bonds formed correctly leading to the properly folded protein. The diselenide bonds dramatically increased stability in physiologically relevant conditions, as confirmed by functional assays.
Many of the protein therapies available now, as well as many more still in development, got their start on computers. Software identifies the protein shapes best suited for therapeutic applications and those designs are tested in a lab. While computational techniques have advanced the design and development of new therapeutic proteins, even the most advanced of these are limited to 20 standard amino acids found in nature that are building blocks of all proteins. Harvard University spinout GRO Biosciences aims to improve protein therapies by expanding this amino acid alphabet.
GRObio has been quietly developing its technology for the past several years. The company has made progress in its preclinical research and it’s now positioning itself to advance its own therapies, and to strike up partnerships with pharmaceutical companies interested in working with the startup’s technology. To support those efforts, GRObio announced on Wednesday a $25 million Series A round of funding co-led by Leaps by Bayer and Redmile Group.
The science behind GRObio comes from the lab of George Church, a Harvard scientist whose discoveries have led to the founding of many life sciences startups. Dan Mandell, GRObio’s co-founder and CEO, was a research fellow in genetics at Harvard, where he worked with Church on computational design of new proteins whose folding and function depends on this new amino acids alphabet, comprised of non-standard amino acids (NSAAs).
Therapeutic proteins are produced by harnessing the protein-translation machinery of bacteria. Companies such as Ginkgo Biosciences, Synlogic, and Absci work with E. coli to produce their commodity chemicals and proteins. For many synthetic biology companies, E. coli are the bacteria of choice because they are inexpensive and easy to use, Mandell said. GRObio also works with an E. coli-based organism. But the company has gone further than what nature provides by recoding the E. coli genome so that these bacteria are able to produce proteins by using NSAAs. GRObio calls these bacteria “genetically recoded organisms,” or GROs.
“What’s special about these organisms is they can make proteins comprised of amino acids beyond the 20 standard amino acids,” Mandell said. “These organisms are the only organisms that can produce these NSAA proteins at high efficiency, and at scale.”
So why would anyone want a GRO-produced protein made from NSAAs? Mandell said that therapeutic proteins made with standard amino acids still have limitations ranging from safety issues to the durability of the treatment. Working with NSAAs enables the production of customized proteins whose shape and chemical properties offer advantages for a biologic drug.
GRObio is working with two families of NSAA chemistries so far. The first, which the company calls DuraLogic, makes a protein that is more stable and improves its half-life. Currently available biologic drugs dosed as frequent injections are inconvenient or undesirable (or both), which leads many patients to miss doses, Mandell said. By making a more stable protein, GRObio could produce a drug whose therapeutic effect lasts for a longer period of time, which means a protein therapy that requires less frequent injections.
The second NSAA family, which GRObio has dubbed ProGly, enables the biotech to directly modulate the immune system, offering a new way to address autoimmune diseases. The way the immune system distinguishes a foreign protein from one that is part of the body is by detecting sugar molecules called glycans on the protein’s surface, Mandell said. GRObio aims to express proteins decorated with human glycans, which would reeducate the immune system to recognize them as belonging to the body.
GRObio hasn’t disclosed what diseases it aims to address, other than to say the technology has applications in autoimmune and metabolic disorders. One of the company’s early projects was a form of insulin modified in a way to enable weekly dosing. The company wasawarded a Phase I Small Business Innovation Research grant in 2019 for that research, followed by a Phase II grant in 2020. Mandell acknowledged that GRObio has worked on insulin, and said the company has received about $1.5 million in non-dilutive capital to support that work.
Without specifying a disease target, Mandell said he expects GRObio could begin its first human tests of a GRO-grown therapeutic protein in 2024. The new financing will support the preclinical research leading up to those tests. GRObio is also looking for pharmaceutical industry partners. Those partners could license GRObio therapeutic candidates, taking on the responsibility of clinical development and potential commercialization of new protein therapies.
Mandell said GRObio is also considering alliances with companies that want to work with NSAAs but can’t because they don’t have access to a production platform that can produce NSAA-based proteins at scale. Mandell said GRObio has been approached by companies that have already designed their own new molecules and are looking at GROs as a way to produce them.
Prior to Wednesday’s funding announcement, GRObio had raised $2.1 million in a 2017 seed financing led by Digitalis Ventures and Innovation Endeavors. Those firms also joined the Series A round, bringing the startup’s total investment to $31.2 million to date.
Genomics and synthetic biology pioneer George Church, PhD, says GRO Biosciences (GRObio), a developer of enhanced protein therapeutics he co-founded based on platform technology discovered in his Harvard Medical School lab, reflects a truism about startup formation.
“Every postdoc has an invention, but not every invention is something that we want to immediately launch,” Church, who heads GRObio’s scientific advisory board, observed recently on “Close to the Edge”, GEN Edge’s video interview series. “We try to incubate them as long as possible inside the lab until we’re sure that they’re mature enough so that we won’t get diluted out immediately by running out of VC [venture capital] money.”
GRO stands for “genomically recoded organisms”—the first production organisms made with modified genomes and engineered protein translational machinery, according to the company.
By recoding the genomes of bacterial strains used in biologics production, GRObio reasons that it can expand beyond the 20 amino-acid building blocks typically found in proteins to introduce non-standard amino acids (NSAAs) that can customize the shape and chemical properties of the protein.
“What we do is to systematically alter the genetic code,” Daniel J. Mandell, PhD, who is GRObio’s CEO, summed up to GEN Edge.
Church’s Harvard Medical School lab first described and characterized GROs in a paper published in 2013 in Science.
In 2015, Church, Mandell and seven co-authors reported in Nature their successful computational redesign of essential enzymes in the first organism possessing an altered genetic code, conferring metabolic dependence on NSAAs for survival.
The following year, Church led a research team in applying recoding to design and synthesize a bacterial genome, an exercise designed to show how new organisms could be created that feature functionality not previously seen in nature.
Also in 2016, Church, Mandell, and four others co-founded GRObio to commercialize the technology by developing protein therapeutics based on computational protein design and synthetic biology technologies. Among the co-founders are Andrew Ellington, PhD, whose lab at the University of Texas at Austin focuses on developing novel genetic codes and synthetic organisms based on engineering the translation apparatus; and Christopher Gregg, PhD, GRObio’s chief scientific officer (CSO).
Mandell was wrapping up a PhD in computational protein design at University of California, San Francisco, about a decade ago, and seeking a postdoctoral position when he came across Church’s research on GROs.
“It was an epiphany”
“For me it was an epiphany: Now we can go beyond the 20 amino acids and build designer proteins that can carry out almost any function. I came to George’s lab to bring these two worlds together of computational protein design and genome-wide recoding,” Mandell recalled.
Daniel J. Mandell, PhD, GRO Biosciences co-founder and CEO
“We did some early work demonstrating that you can in fact predictably engineer proteins whose folding and function depend upon non-standard amino acids, to convince ourselves that we really can do this in a rational way. That’s when we turned our attention to the question of what are the outstanding problems in the clinic that we might address using that technology.”
Mandell joined Church’s lab within a year of Gregg joining: “We very quickly realized we both had entrepreneurial designs and mesh very well, and we put a lot of time into thinking about how we could try to commercialize this technology.”
Gregg, now GRObio’s CSO, was pursuing a PhD in glycobiology, studying how glycans interact with the immune system. He started a short-lived synthetic biology startup focused on biofuel, before switching gears to integrating synthetic biology with organism and genome design.
“Luckily, I was able to get George’s interest in my thesis, which had to do with glycobiology and human-specific disease preponderances,” Gregg recalled. “I came in as the organism [GRO] was getting finished. It was just such a ripe platform for trying new things and solving new problems. Then when Dan and I realized that we had the same interests, we just started running with it.”
That pursuit paid off for GRObio last month, when it completed a $25-million Series A financing co-led by Leaps by Bayer and Redmile Group. Redmile is a San Francisco venture and private equity investment firm. Leaps is the equity investment arm created by Bayer to establish new companies and invest in early-stage technologies with breakthrough potential to “fundamentally change the world for the better.”
Bayer came to invest in GRObio, Mandell said, through relationships he had with the pharma/consumer goods/agbio giant and contacts from GRObio’s network of investors. Among investors joining the financing were Digitalis Ventures and Innovation Endeavors. (The Series A brings total investment in GRObio to $31.2 million so far.)
Proceeds from the financing, GRObio said, will support development of its GRO platform, a scale-up of bioprocess manufacturing, preclinical validation studies, and IND-enabling studies for GRObio’s pipeline of NSAA protein therapeutics designed to treat autoimmune and metabolic diseases.
“These are just initial focus areas,” says Mandell. “Autoimmune disease and metabolic diseases are a really a small part right of this broader universe [of opportunities]. But there are specific indications in there that we’re focusing on.” The company isn’t yet disclosing those indications, except to say they will address unmet clinical needs.
Beyond the pipeline
Taking this universe of NSAA chemistries, Mandell and colleagues want to ask some big questions. “Which problems really can’t be solved in the clinic without this new expanded universe of chemistries at the amino acid level? Some of these problems have interesting-looking solutions coming down the clinical development pipeline,” Mandell said. “That’s not really where we want to play. We want to play in areas where we think we can solve Holy Grail challenges and there isn’t another way to go about this.”
GRObio has constructed a “biofoundry” consisting of proprietary computational protein design and robotics pipelines, with the aim of streamlining development of NSAA translational machinery and NSAA protein products. The biofoundry applies strain and genome engineering, automation, analytics, and protein design software to build uniquely scalable NSAA protein “factories” from trillions of candidates.
GRObio emerged from stealth mode in 2017, raising $2.1 million in seed funding led by Digitalis Ventures, with participation from Innovation Endeavors, the venture capital firm whose co-founders include Eric Schmidt, Google’s former CEO and later executive chairman of Google and its parent company, Alphabet Inc.
From three people when it started, GRObio has since grown its staff to 16 people. “We will double in size by 2023,” Mandell said.
GRObio hopes its alphabet-expanding approach will enable it to grow its own significant share of a protein therapeutics market that according to Market Research Future (MRFR) is expected to increase over the next six years at a compound annual growth rate of 6.86%–for a nearly 60% rise to about $290.69 billion in 2027 from $182.69 billion this year.
GROs are intended for high-efficiency, commercial-scale production of proteins with NSAAs. These NSAAs are intended to enhance protein therapies with capabilities such as unprecedented duration of action and more precise control of the immune system.
GRObio is building its pipeline by advancing its first two product families of NSAA platform chemistries: DuraLogic™ is designed to enable flatter pharmacodynamic profiles and relaxed dosing schedules, while ProGly™—short for “programmable glycosylation”—are designed to produce biologics that enable the immune system to treat autoimmune disease, or to eliminate immunogenic side effects of protein-based therapies.
DuraLogic integrates NSAAs to enhance and maintain the three-dimensional structure of proteins needed for therapeutic activity. “That means things like resistance to proteases that can degrade the therapeutic, resistance to reducing agents that can break bonds in the protein that will render it inactive or misfolded, and the potential in some cases to increase the thermostability of the protein, so it can either last longer in storage, or just have a longer in vivo half-life,” Mandell said.
ProGly consists of glycan-containing NSAAs designed to induce or inhibit an immune reaction. GRObio says its GRO platform enables precise placement of defined ProGly compositions on the protein surface needed to elicit immune response.
Re-educating the immune system
“You can actually retrain or re-educate the immune system to treat a particular protein as self or non-self by administering that protein with a particular glycan’s signature. You give it what’s called a tolerizing signature,” Mandell explained. “The body will remember that this is a self-protein, and over a short period of time can begin to actually reverse the autoimmune response to that protein.”
GRObio expresses that protein in its platform, decorated with ProGly NSAAs that take on the form of human-like glycans.
“By putting that into your body, your immune system begins to learn that this is a self-protein and it builds up an immune memory of that protein and stops attacking it,” Mandell said. “This is really a way that we can begin to reverse a number of different autoimmune diseases, by taking an antigen-specific approach.”
That approach, he added, contrasts with broadly immunosuppressive drugs that turn down auto immune response by knocking down a patient’s immune system—an approach Mandell said increases susceptibility to infection and cancer and also often causing metabolic disorders: “What we want to do is re-educate the immune system to treat the one protein or the small number of proteins that you’re reacting to as self-proteins for the first time.”
The genetic code of each gene combines the four natural nucleotides of DNA—adenine (A), cytosine (C), guanine (G), and thymine (T)—to spell out three-letter codons specifying an individual amino acid. There are 64 different naturally occurring codons that encode 20 natural amino acids.
“What we did was to modify the genome to remove all the instances of one or more of those codons. Then, by installing on what we call new translational machinery from the cell that recognizes a particular codon, we can actually now reassign the meaning of part of the genetic code to direct the incorporation of a new amino acid,” Mandell said.
The company’s GRO platform expands the amino-acid alphabet to overcome limitations of protein therapeutics to address product stability, immunogenicity, and delivery into the cell.
Exploiting redundancies
“We’re exploiting redundancies in the genetic code to reassign redundant codons to new, non-standard amino acids, because there’s more than one way to specify a particular amino acid or stop,” Mandell explained.
For example, three codons code for the “stop” signal (UAG, UAA, and UGA), halting the elongation of a growing peptide chain. GRObio developed a recoded strain of bacteria that replaces all UAG stop codons with UAA stop codons and reprogrammed the UAG codon to new amino acids such as selenocysteine, a naturally occurring amino acid found in the cell, yet rarely incorporated into proteins.
By replacing all codons that code for cysteine residues in a protein with UAG, selenocysteine can be selectively incorporated in place of cysteine residues to form stabilizing diselenide bonds. Proteins stabilized with diselenides maintain stability and resist the reduction found both in human blood plasma, which destabilizes therapeutics, as well as in solvents and buffers, which destabilize industrial enzymes.
“It was quite evident that people were excited about working with the first recoded strain and they started looking around for what the best products were,” Church recalled. “The first thing we had published on the first NSAA we’ve published for biocontainment was bipA [biphenylalanine], and that did not seem like a good starting product. But diselenides, which we did in collaboration with Andy Ellington’s lab, looked like a great initial product, and that was quite enough to get the company launched.”
Gregg cited insulin as an example of a treatment that could be engineered for less frequent dosing through a selenocysteine NSAA that could be designed to prevent the breaking of bonds between the peptide chains that renders the molecule non-functional.
“Now all of a sudden,” he said, “you’ve got a fundamental mechanism by which you can say, this NSAA will support the survival of this molecule in this environment, on a timescale that we think will be beneficial to patients as a product.”
Synthetic biology has brought many breakthroughs to the biotherapeutics space over the last decade. The dropping cost of sequencing and precision genome editing has paved the way for personalized medicine. At the same time, generative AI technology has enabled the designing of antibodies with a much higher clinical success rate. Yet, scientists’ ingenuity is still challenged by the laws of biology. All biologics are susceptible to unpredictable degradation rates and immune responses, in addition to being constrained to the 20 natural amino acids that make up these therapeutics.
But now, one company is challenging that paradigm. Pearl Bio, a synthetic biology company backed by Khosla Ventures, has recently emerged from stealth mode with a hefty IP portfolio of 24 patents that protect their groundbreaking technology: a genetically recoded organism. With it, Pearl Bio is creating entirely new classes of materials for smart biologics.
Pearl’s genetically recoded organism, combined with the engineered translational machinery of the cell, breaks the rules of life by enabling the incorporation of building blocks beyond the 20 amino acids that exist in nature. This technology can disrupt the medical paradigm by producing novel chemistries that solve existing challenges in the immunotherapeutics space and pave the way to completely new materials.
What is a Genetically Recoded Organism (GRO)?
Pearl’s cornerstone technology is a genetically recoded organism (GRO), which heralds a new era of synthetic biology. The rules of life are written in a four-letter genetic code: A, T, C, and G. These letters form three-letter words—called codons—that encode the amino acids which make up the tissues and enzymes of all living beings. However, biology is not perfect: there are 64 possible combinations of ATCG but only 20 amino acids. This phenomenon is known as “redundancy.”
The idea of using those redundant codons for a practical purpose has been around for a while. “It dates back to the late 2000s. I was working as a postdoc in George Church’s lab, together with Michael Jewett,” recalls Farren Isaacs, Co-founder and Science Advisor at Pearl Bio. “We were always the first people to get to the lab in the morning. There was no one around at that time, and we would brainstorm ideas.”
Isaacs and Jewett, Pearl Bio’s other Co-founder and Science Advisor, were working on the bleeding edge of synthetic biology at that time: “I was recoding genomes, and Michael was engineering ribosomes. We realized that when you put those two technologies together, you have the ability to basically repurpose the genetic code and the translational machinery of any cell to produce entirely new materials,” says Isaacs.
In 2013, scientists from the Isaacs lab managed to free up one of those three-letter combinations by editing the entire E. coli genome. Now the missing codon could be assigned to code for anything else, such as non-natural amino acids, which do not occur in any living beings. By introducing these new-to-nature building blocks, you could make proteins that are impervious to degradation, target specific tissues and disease states,and attach highly specialized payloads.
“That could lead to fundamentally new kinds of therapeutics that have longer stability and reduced immunogenicity,” says Isaacs.
A New Way to GRO
Years went by, but Isaacs, Church, and Jewett kept working on tweaking the technology to improve and expand on what it could do. In 2020, the two decided to co-founder Pearl Bio together with Amy Cayne Schwartz, Chief Operating Officer & Chief Business Officer, and George Church and Jesse Rinehart of Yale as Science Advisors. Pearl Bio was officially financed and launched in Q3 of 2021.
“This is something that we’ve been incubating over many years: developing the technology, de-risking proof of concept, and filing IP,” says Isaacs. “With platform technology companies, you want to be poised to advance product development from day one.”
And Pearl Bio was. They had been working to secure an impressive portfolio of 19 patents to corner the market of multi-functionalized biologics. The newly issued U.S. patent 11,649,446 gives Pearl Bio the exclusive license for engineering programmable biologics by encoding synthetic chemistries and now brings their total patent figures to 24.
When the paper describing the first GRO was published in 2013, the technology and the strain were released publicly. But just like the first version of iOS, there were many things that needed to be improved to make the strain better suited for commercial applications. For example, the initially published version only had one codon freed up, which meant that it could only encode one alternative amino acid. Pearl took this concept further and now also holds exclusive rights to a newly developed GRO with two open coding channels to endow multi-functionality into protein therapeutics.
“Another key piece of IP is the orthogonal tethered ribosomes. This allows Pearl to engineer the ribosome to work efficiently with exotic substrates beyond L-alpha amino acids, opening access to new classes of therapeutic biomaterials. This capability holds promise to transform biopharmaceutical development,” says Jewett.
Other companies have also taken a stab at challenging the paradigm within the biotherapeutics space. For example,Ambrx, a clinical-stage biopharmaceutical company using an expanded genetic code technology, IPO’d for $126 million in 2021. However, Ambrx’s technology does not use genetically recoded organisms. Synthorx is another synthetic genome company in this space which was acquired by Sanofi for $2.5 billion in 2019. Their Expanded Genetic Alphabet technology that adds a novel DNA base pair can be used to create optimized biologics. GRO Biosciences, a company in Cambridge, MA, is expanding the amino acid alphabet to deliver on the promise of protein-based therapies. Their platform comprises Genomically Recoded Organisms (GROs) for high-efficiency production of non-standard amino acid (NSAA) proteins at commercial scale.
What distinguishes Pearl Bio is that they hold a number of broad-blocking patents giving Pearl the exclusive right to encode synthetic amino acids using engineered synthetases and translation machinery to drive multi-site incorporation of synthetic amino acids and other building blocks with site-specificity at high yield and purity.
Schwartz thinks this technology is poised to overcome a lot of the shortcomings of current biologics on the market: “For example, optimizing the drug-antibody ratio has been a defining challenge because when you attach the drug to natural amino acid residues, you are limited in the number and specific location since multiple lysines, for example, are found in a given protein and are critical for its function. Thus, approaches that attach to natural amino acids are constrained, lead to heterogeneous products, and typically ablate protein function. By contrast, Pearl can attach up to 50 synthetic amino acids at monomeric precision to tune therapeutic properties while preserving protein sequence and function.”
Such precision enables the programmability of a therapeutic window with a half-life of up to three weeks to address specific disease indications or patient populations. “With this, we have the opportunity to advance both best-in-class and first-in-class therapeutics to address critical unmet needs and solve challenges plaguing the biologics industry,” says Schwartz.
The Future of Biotherapeutics
Pearl has positioned itself as the leader in this new field thanks to advancing technology and staking the IP landscape. They have developed exclusive next-generation multifunctional capabilities, such as adding synthetic monomers, multiple types of modifications, and multiple locations.
“It’s exciting to see ideas from whiteboards over 15 years ago realized in technologies that are now poised to transform the therapeutic landscape and evolve novel biomaterials. Pearl is pioneering a new era of biotherapeutic design by enabling access to new disease targets and evolution of entirely new classes of molecules,” says Church.
“Today, we can access two distinct functionalities. With our Series A funds, we will advance the technology to access three or more distinct functionalities,” says Schwartz. “We are excited to drive next-generation molecules to the clinic to change the therapeutic landscape with the evolution of smart programmable biologics.”
Pearl Bio, a synthetic biology company backed by Khosla Ventures, is recoding life to create a new era of biologics and biomaterials. A newly issued breakthrough U.S. patent 11,649,446 related to engineering programmable biologics by encoding synthetic chemistries bolsters Pearl’s patent portfolio to corner the market of multi-functionalized biologics with Pearl’s exclusive license to the issued patent. The Pearl team includes world leaders in synthetic biology, genome recoding, and ribosome engineering: Drs. George Church (Pearl Bio, Scientific Advisory Board), Farren Isaacs (Yale University), Michael Jewett (Stanford University) and Jesse Rinehart (Yale University).
“By encoding diverse synthetic chemistries into proteins, Pearl is able to tune half-life, target delivery to diseased cells, and attach cytotoxic payloads to tailor valuable therapeutic properties, overcoming key barriers preventing market approval,” explained Co-Founder Amy Cayne Schwartz. Pearl’s platform leverages 24 exclusively licensed patents and applications evolved over the last decade by the company’s scientific Co-Founders, Dr. Isaacs and Dr. Jewett. The company is advancing partnerships with pharmaceutical companies alongside internal programs to develop next-generation “smart” biologics.
Bringing together the newly-issued patent with existing broad blocking patents on genomically recoded organisms, tethered ribosomes and engineered translational machinery enables access to new frontiers by site-specifically encoding synthetic monomers to derive novel biologics and biomaterials.
Pearl’s technology preserves the natural protein activity while endowing valuable therapeutic properties to address defining challenges in biologic drug development – toxicity, stability, and targeted delivery – fast-tracking the path to market. For example, compounds designed to sustain presence of a cytotoxic payload in the tumor microenvironment coupled with access to novel targets will open-up entirely new therapeutic opportunities to address unmet medical needs and transform patient quality of life.
About Pearl Bio
Backed by Khosla Ventures, Pearl Bio was launched by Scientific Co-Founders Drs. Farren Isaacs (Yale), Michael Jewett (Stanford), and Amy Cayne Schwartz, J.D. (Pearl Bio) bringing together 24 patents in a platform technology to advance multi-functionalized biologics and biomaterials by encoding synthetic chemistries. Broad blocking patents afford freedom to operate, and the company has rapidly advanced capabilities in-house and through pharmaceutical partnerships. Pearl Bio may be followed at: pearlbio.com Twitter: https://twitter.com/PearlBio
20 June 2023. A new U.S. patent, licensed to a start-up biotechnology company, describes processes for linking together chains of synthetic peptides into therapeutic proteins. The U.S. Patent and Trademark Office issued patent number 11,649,446 last month to four inventors at Yale University and elsewhere, including a founder of the company Pearl Bio in Cambridge, Mass. that acquired rights to the technology.
Pearl Bio is a two year-old enterprise discovering synthetic combinations of peptides, short chains of amino acids, for new therapies and bio-based materials. The company is commercializing research by biomedical engineering labs of its scientific founders Farren Isaacs at Yale University and Michael Jewett at Stanford University, as well as Yale physiology professor Jesse Rinehart and Harvard Medical School geneticist George Church. Pearl Bio says the new patent is the latest of 24 patents it licenses exclusively for its basic technology.
The Farren Isaacs lab at Yale studies genome engineering techniques, particularly for high-volume programming of genetic chemistries in single cells and across cell populations. The lab says its discoveries make possible large-scale assembly of genomes into hierarchies that express genetic modifications to achieve desired outcomes, even new organisms if needed. This capability includes engineered protein synthesis in the ribosome, where messenger RNA translates and sequences genetic codes into chains of amino acids to form peptides, then linking together peptides into longer multiple peptide chains and proteins.
Produce synthetic proteins in greater yields
The new patent, which lists Isaacs as the lead inventor and assigned to Yale University, describes processes for preparing multiple peptide chains from amino acids, particularly amino acids not normally found linked together in their natural states. The patent includes methods for producing synthetic proteins in greater yields than conventional processes, or where conventional techniques would not allow for those combinations of amino acids to produce adequate yields or purity, or even occur in some cases.
“By encoding diverse synthetic chemistries into proteins,” says Pearl Bio co-founder and chief operating officer Amy Cayne Schwartz in a company statement released through BusinessWire, “Pearl is able to tune half-life, target delivery to diseased cells, and attach cytotoxic payloads to tailor valuable therapeutic properties, overcoming key barriers preventing market approval.”
Pearl Bio says its synthetic proteins retain their natural protein activity, but still allow for tuning the protein chemistry to reach specific targets, maintain stability, and reduce toxicity. The company cites as examples delivery of cancer-killing proteins to tumors through the protective microenvironment, while also accessing novel therapeutic targets. Pearl Bio says it’s forming partnerships with drug makers to develop what the company calls the next generation of smart biologics. In addition, says the company, its process makes possible genetically altered organisms that can produce strings of basic components constructed into wholly new biologics and bio-based materials.
Merck & Co. has signed up to Pearl Bio’s synthetic biology platform, offering up to $1 billion in biobucks for potentially cancer-busting biologics.
The collaboration could pile more biologics onto Merck’s pipeline and serves as validation for Pearl, which emerged from stealth in June 2023 with the backing of Khosla Ventures. Merck is paying an undisclosed upfront fee and offering up to $1 billion in milestone payments for Pearl to identify biologics to treat cancer.
The biotech describes itself as creating “template-directed biomaterials with tunable properties.” Co-founder, Chief Operating Officer and Chief Business Officer Amy Cayne Schwartz elaborated in an email to Fierce Biotech, describing how the platform can “encode synthetic chemistries to tune therapeutic properties (eg half-life, drug antibody ratio.” The company is in the process of raising its series A round, she told Fierce.
“Pearl Bio is recoding life for a new era of programmable or ‘smart’ biologics,” Schwartz wrote. “Series A resources will be used over the next two years to drive molecules to the clinic and further differentiate our technology with the ability to endow three distinct functionalities with tunable properties into biologics.”
Pearl was spun out of research from Farren Isaacs, Ph.D., and Michael Jewett, Ph.D., that centers on using ”genomically recoded organisms” to add synthetic chemistry to biologics. The company counts famed molecular engineer and serial entrepreneur George Church, Ph.D., as one of its scientific advisers. In June 2023, the U.S. granted a patent to Pearl and its founders that bolsters their multifunctional biologic ambitions, cementing future use of tools like synthetic amino acids and tethered ribosomes.
As for Merck, it’s just the latest collab to land on its conveyor belt of licensing pacts. The Big Pharma’s recent slate of bets has largely focused on oncology, including deals with C4 Therapeutics and Daichii Sankyo, alongside the $680 million acquisition of Harpoon Therapeutics.
Juan Alvarez, Merck’s vice president of discovery biologics at Merck Research Labs, described Pearl in a release this morning as a “pioneer in developing recoded organisms.”
Farren Issacs Lab at Yale University 에 보여진 연구내용을 보면 MAGE (Multiplex Automated Genome Engineering)과 CAGE (Conjugative Assembly Genome Engineering) 을 통해서 Recorded E. Coli 를 만들 수 있습니다. Hierarchical assembly of codon mutations를 통해 다양한 사이트에 정확도 높은 조작을 할 수 있다는 설명입니다.
(Picture: Bruce Booth, Ph.D., Partner at Atlas Ventures)
안녕하세요 보스턴 임박사입니다.
주식투자에서 Option Investing이라는 방식이 있습니다. 이 방식은 Structured Deal로서 Downside Risk를 줄이고 대신 Upside Gain도 어느 정도 포기함으로써 어려운 시장에서 살아남은 방법이라고 할 수 있는 Venture Investing Strategy 중에 하나입니다. 보스턴의 벤처캐피탈인 Atlas Ventures의 Bruce Booth 박사는 “Option-to-Buy M&A” Model을 가장 먼저 시작한 VC로 기억합니다. 2011년에 시작한 이래 아래와 같은 다양한 회사들이 이 모델에 의해 투자 회수가 되었습니다. 최근은 IPO시장이 2010년대에 비해서는 훨씬 좋은 상황이지만 언제든지 시장은 반대로 돌아설 수 있다는 생각으로 이 모델에 대해 다시 한번 생각을 해 보고자 합니다.
2007-2008년에 미국 서브프라임모기지 사태와 리먼-브라더스 사태로 시작된 글로벌 금융위기는 전세계 주식시장을 비롯한 금융시장에 오랜기간 충격을 주었는데 바이오텍의 충격은 당시 매우 심했습니다. IPO 시장은 수년간 적은 수의 기업만 가능했을 뿐만 아니라 IPO Valuation도 낮아서 당시 Venture Capital 에는 자금 회수를 할 방법이 상대적으로 어려운 실정이있습니다.
빅파마를 비롯한 바이오텍의 대량 해고가 매년 끊이지 않았던 어려운 시기였고 따라서 빅파마들의 파이프라인의 생산성도 크게 낮아지고 있었습니다. Original Drug Patent Expiry에 의한 Generic 압력은 그 어느 때보다 높았습니다. 반면 미국 대학이나 연구소에서 생산되는 기술혁신은 새로운 Drug Targets와 Platform이 태어날 기회가 되기도 했습니다.
2010년까지 미국 IPO 시장에 대한 Atlas Ventures의 블로그 글이 당시 상황을 잘 말해 줍니다. 2010년은 Nasdaq 시장지수를 반영한 바이오텍 IPO에서 전년에 비해 5% 정도 상승함으로써 그나마 선방한 해였습니다. 하지만 바이오 기업의 특성 상 나스닥 시장에서 가격이 중요한데 여전히 낮은 수준의 가격으로 벤처캐피탈의 입장이나 Crossover Investors 입장에서도 좋은 상황은 못되었던 것 같습니다.
Several recent stories from WSJ and VentureWire have highlighted the challenging performance of the IPO markets for biotech in 2011. It has indeed been tough: more shares offered at lower prices = more painful dilution. From a pricing perspective, the Class of 2010’s thirteen biotech IPOs faced similar challenges.
Surprisingly, however, the markets have been reasonably good to the 2010 class since their IPOs. Here is the price performance relative to their IPO price as of today:
The average and median performance of this “Class” is 18% and 14%, respectively – which is quite abit better than several of the recent prior classes performance. Ventrus, Aegerion, AVEO, Anacor have all appreciated by more than 30% since their IPO.
However, the NASDAQ itself has also been on a tear, up above 30% since mid-2010. To get a sense for individual company outperformance vs the market, I’ve adjusted the performance of the Class by the NASDAQ’s performance from the individual IPO dates:
The order shifts as one would expect in a bullish stock market with newer IPOs moving up in the ranking (less adjustment) and older IPOs moving down (more adjustment). Importantly, however, the class average stock performance was still up 5% after adjusting for NASDAQ market performance. That’s respectable.
From a venture standpoint, since IPOs are financings not exits, understanding the price performance relative to their last private round is important. From what I can tell, it has actually been pretty strong. Here’s a snapshot of performance of 12 of these 13 where I could get the last private round pricing (courtesy of a friendly biotech investment banker). Unfortunately I don’t have data on Ventrus Biosciences. I’ve plotted the current stock price on the Y-axis and implied “last round” pre-IPO price on the X-axis. Any ticker above the line is at least “in the money” for the last private investor (maybe or maybe not for the early investors depending on the step-ups or cramdowns along the way); below the line are “underwater” positions for that last round. Good news is most are near or above the line – with considerable outperformance for Aegerion (nearly 3x) and Complete Genomics (2x). Tengion is sadly quite an underformer – roughly 10 cents on the dollar.
The takeaway message here is that despite the ‘doom & gloom’ around biotech IPOs, there’s some glimmer of hope in the post-market performance for the “Class of 2010”.
Hopefully the pricing struggles of Class of 2011 will be forgotten with some strong stock performance ahead.
이런 어려운 금융환경 속에서 Atlas Ventures는 Bill Gates와 함께 Nimbus Discovery LLC를 만드는 새로운 시도를 합니다. Nimbus Discovery LLC는 Option-to-buy M&A Model의 선구자적 기업이라고 볼 수 있습니다. 2009년 당시 아직 비상장기업이었던 Schrödinger라는 Computation drug discovery company가 있었는데 Bill Gates도 이 회사의 대주주 중 한명이었습니다. 당시 Schrödinger가 WaterMap이라는 In silico SAR Model을 개발했는데 이를 이용해서 Global Virtual Biotech을 만드는 실험을 한 것입니다.
Nimbus Discovery LLC는 Virtual Biotech Model을 Computational Drug Discovery에 연결한 방식이었습니다. 2009년부터 2010년까지 1년간 인더스트리의 백여개 표적을 조사하고 그 중에서 가장 경쟁력이 있다고 판단하는 십여개의 표적에 집중해서 Project 단위별 C-Corp를 만들고 IP를 각 회사에 집중시키는 방식이었습니다. Schrödinger의 60명의 박사들이 참여하고 해외 CRO들이 합성, Biology, DMPK 등을 하는 방식으로 해서 실제로 Nimbus의 인력은 필요 인력의 10-15분의 일에 지나지 않게 만든 것입니다.
Bill Gates has just backed one our new startups – Nimbus Discovery LLC – as part of an extension to the seed tranche. Here’s the press release.
It might come as a surprise to some, but Bill Gates has been a long-time biotech supporter: he was a founding investor in ICOS and was on that Board for 15 years, and importantly, he’s also one of the largest investors in Schrödinger, the world’s leading computation drug discovery company, and our founding partner with Nimbus.
So, with this financing, we’re launching Nimbus out of ‘stealth mode’. Here’s the story.
We founded Nimbus Discovery in 2009 with Schrödinger and have incubated the company here at Atlas for the past couple years. It’s fair to say Nimbus is a rather unconventional biotech, possessing three distinctive features.
Unique Drug Discovery Partnership with Schrödinger provides access to an unparalleled technology suite without the financial burden of having to build it organically. Beyond the sheer breadth of Schrodinger’s software offerings, the crown jewel from our perspective is their new technology for evaluating the energetics of specific water molecules in binding sites called WaterMap™.
Our bodies are 90+% water and yet most structure-based drug design models fail to integrate proper solvent (water) effects with regards to both entropy and enthalpy. WaterMap™ does this. We’ve already found it to be an incredibly powerful tool for identifying specific optimization strategies based on novel water-energy-driven Structure-Activity-Relationships. WaterMap™ is a incredibly well validated technology and has been applied (retrospectively) to about 45 different targets using ligands that have highly heterogenous structures. Not only does WaterMap™ accurately predict binding affinities, it explains SAR that would otherwise be inexplicable. In short, super cool science at the cutting edge of in silico drug discovery.
Our partnership with Schrödinger provides us with far more than just this software package though – we get a large number of dedicated computational chemists, access to thousands of processors via their cloud computing network, new unreleased software algorithms, and exclusivity around specific targets. Continuous, advanced access to the most cutting edge technology applied in a personalized way to our targets allows Nimbus to maintain its first mover advantage.
Moreover, Schrodinger continues to make a huge investment in its platform leveraging an army of ~60+ PhDs and the deep-pocketed support of Bill Gates and David Shaw. (As an aside, it’s probably the only biotech with two billionaires as its top two investors). It is no surprise that Schrodinger has led innovation in the field: the company currently has 50+ peer reviewed publications many of which are among the most heavily cited articles in the in silico modeling space.
Ultra-lean “virtually integrated, globally distributed” R&D operating model to aim for exquisite capital efficiency. We’re really pushing the envelope on virtual drug discovery. We have 10-15x more FTEs working for the company externally as inside the company.
The core team is two incredible individuals (Rosana Kapeller and Jonathan Montagu) who, in addition to being very smart seasoned biotech startup veterans, excel at integrating remote workstreams and collaborators. We’ve got several teams at offshore CRO partners doing biology, chemistry, crystallography, in vivo work, etc…. Not to mention a core set of KOL academic collaborators. We think we’ll be able to work on 3-4 programs with this setup.
It’s paying dividends already: on less than $2M spent, we’ve generated a selective, potent IRAK4 inhibitor (cancer, inflamm) and a set of lead scaffolds against other targets.
One of the key features of the operating model is the virtual integration of all these pieces, with in silico model refinement in the core of the ‘engine room’ so to speak. WaterMap and tools like it depend on constant structural model enhancement, which requires real time integration of project data into our models. Our remote virtual teams interact on almost a daily basis to integrate these new streams of information. This allows us to use these tools for more than just improving selectivity and potency – but also to more precisely know which part of molecules to optimize for PK/ADME concerns as well.
Novel asset-centric corporate structure to promote liquidity and capital velocity. Back in 2009 we spent a lot of time figuring out how to leave the limitations of the traditional C-corp structure behind and adopt a more flexible LLC-holding company structure with target-specific subsidiaries as C-corps. A few companies have recently announced they are doing this as well, like Adimab and Ablexis. This structure does a couple very valuable things (beyond creating a job for accountants to track the project financing).
First, it enables project-driven ‘clean’ transactions with Pharma, such that a Pharma can just acquire the target-specific subsidiary and own the IP/assets of a particular program if it so chooses.
Second, and importantly, it solves the classic drug discovery illiquidity problem (where it takes 7-10 years to get liquid via M&A or IPO); this LLC structure enables us as shareholders to cycle capital back to our investors in a tax-efficient manner on a per project basis. Furthermore, it creates this type of ‘deal optionality’ without foregoing any of the traditional benefits of C-corps.
At the end of the day these three elements are great value enablers, but ultimately it’s about the medicines we discover. To pick the targets we sought to generate drugs against, we took an orthogonal approach to traditional “biology” driven target selection. History has shown that generally in silico technologies in drug discovery are helpful tools for the vast majority of targets, but not game-changing. With Nimbus, we wanted to let the technology identify targets where its new insight into SAR was potentially transformational rather than incremental – essentially, to find the rare 10% of targets where these technologies offered a compelling path to new, differentiated chemical matter against hot targets. To accomplish this, we spent the first 12 months of the company screening several hundred ‘hot targets’ in the industry’ pipeline before picking the 10% or so to focus on with Schrödinger.
Our two lead programs today:
IRAK4. One of the most interesting immune-kinase targets in both B-cell cancers (like the IRAK4-dependent MyD88-driven Activated Diffuse B-cell lymphoma and inflammation. It’s traditionally been very challenging to get selectivity and cellular potency; we’ve managed to generate a series that addresses both of those and aim for a development candidate by end of 2011.
ACC or Acetyl-CoA Carboxylase. A critical enzyme in the metabolism of lipids and a top target for obesity as well as cancer metabolism. It’s been very hard to drug effectively. We’ve managed to get very interesting leads against a unique allosteric domain that should enable a differentiated profile.
Saving the best for last, it’s fair to say our team is exceptional. Rosana Kapeller is our CSO and was founder of Aileron Therapeutics after nearly a decade at Millenium. Jonathan Montagu is our CBO and was with Concert, J&J, Chiron and BCG. The broader team of folks at Schrodinger, like Ramy Farid (President of Schrodinger and founding Board Director of Nimbus), and our chemistry leadership (Ron Wester, Gerry Harriman, Donna Romero, John McCall) are also incredible.
With that rundown, we’re officially out of stealth mode and aiming to close on a Series A soon. Exciting times.
Nimbus의 Series A를 한지 2년 후 (창업 후 4년 후) 이 모델에 대한 블로그 포스팅이 있었는데 당시는 ACC 프로그램이 DC selection을 한 상태였습니다. Investor 입장에서 Platform Company인 Nimbus Discovery LLC는 영원히 운영되고 자회사들만이 독자적으로 생존하며 매각되는 조건의 구조였습니다.
A couple years ago we unveiled a new startup called Nimbus Discovery LLC which was experimenting with a new model that combined three key elements: Schrodinger’s cutting edge in silico drug screening and design platform, a truly virtual and globally distributed operating model for drug discovery, and an asset-centric LLC-based corporate structure (discussed here).
Although it’s too early to tell what eventual value will be created from this experiment, the company’s early biomarkers are strongly positive. Nimbus has cracked two very tough-to-drug targets of high interest to Pharma (immunokinase IRAK4 and lipid-pathway regulator ACC), and is entering IND-enabling development this year. The technology and virtual operating model have worked well together in efficiently delivering high quality drug candidates.
Importantly, the market validation of the model has also been positive: today Nimbus announced a deal with Monsanto, and last month announced a similar deal with Shire – both involve collaborative drug discovery with a pre-defined path to liquidity around those projects. Given the unique nature of the deals, I thought it would be worth sharing more details and a few general reflections on the model.
Both deals are structured to take advantage of the Nimbus asset-centric approach: they involve equity or equity-like investments in individual R&D programs housed in standalone subsidiaries, alongside an option to acquire those subsidiaries at a specific milestone with pre-negotiated deal economics. These are very enabling for Nimbus: project-based resourcing to support the prosecution of a pipeline with clear value creation points defined at the outset without the need for dilutive funding at the parent LLC level.
These collaborative deals were born out of close relationships Atlas has with both Monsanto and Shire. Over the past few years, both companies have been able to watch Nimbus deliver against its existing programs, in particular for ACC, IRAK4, Tyk2. Here’s the short promotional on their programs’ progress On ACC, it took Nimbus only 16 months from standing start with a virtual screen to get to a fully characterized Development Candidate (DC) with a first-in-class allosteric regulator of the target; for IRAK4, the team has discovered truly selective inhibitors with potent in vivo activity and DC-like profiles; and lastly, they have cracked the Tyk2 selectivity challenge vs closely homologous JAK2 and other JAK family members. The progress of these case studies and the familiarity they had with our team definitely facilitated both transactions. More evidence for why tighter collaborative Pharma/Venture relationships are value-creating.
The bigger picture: why these deal structures make sense
For the biotech, these deals help build a portfolio comprising multiple program-focused entities under an LLC umbrella. In some respects, the pipeline becomes a collection of call options on individual paths of potential liquidity.
For Pharma, these structures can be tailored to the requirements and sensitivities of each partner, in many cases enabling what could be described as a P&L-sparing, “balance sheet supported” portfolio of innovative projects. This may not always be the interest of a partner, but accessing the otherwise inaccessible cash on the balance sheets of Big Pharma is a definite positive for these deal structures.
For shareholders, including investors and team members, this model secures potential routes to liquidity that accrue as programs are progressed and monetized through development – importantly without having to sell the entire company. In essence this model creates the evergreen drug discovery stage biotech – a real unicorn in the history of biotech (because most drug discovery biotechs have to either sell or become later stage development players to achieve liquidity).
Lastly, the structure has enormous financing flexibility: any individual subsidiary/program can be financed separately if desired – creating options for going longer on specific programs without diluting the parent platform company (or for a new investor, without having to fund drug discovery if that’s not their interest).
Nimbus certainly anticipates doing more of these types of structured transactions, both for its lead programs (IRAK4, ACC, Tyk2) and de novo collaborations around jointly-identified targets. Several of our other platform-based drug discovery companies, like RaNA Therapeutics, are structured in this way and will likely be pursuing deals of this type. Other drug discovery platform biotechs, like Forma and Viamet, have also been experimenting with versions of this LLC-holding company model. Several subsidiary-level deals have been done across the industry (like Forma-Genentech, among others). To my knowledge, none of these have yet to hit their acquisition-triggering milestones. It will be exciting to see what happens when this crop of deals matures to their pre-defined endpoints.
Creatively thinking about new approaches, new business models is part of innovating around the venture model – some experiments will work, some won’t. But the Nimbus experiment feels pretty good right now.
그로부터 10년이 흐른 후 Atlas Ventures의 Jeb Keiper가 14년간의 Nimbus Discovery LLC Model에 대한 이야기를 잘 정리해 주었습니다. 3개의 Chapter로 구성이 된 “The Book of Nimbus”라는 책입니다.
Nimbus Therapeutics began 14 years ago. The premise at the time was that putting computational chemistry in the primary position for new molecule ideation would upend the drug discovery paradigm. It did just that. Three best-in-class molecules in the clinic, over $400 million invested and over $4 billion returned to equity holders, all while focused on our mission to design breakthrough medicines for patients.
Fourteen years on, this corporate experiment has gone far beyond the initial idea, and has established an R&D engine more effective than most big pharma R&D groups at producing best-in-class small molecules against targets that matter in human disease biology. Throughout that time Nimbus has not just built functional capabilities and continued adopting technological innovation, but importantly has worked tirelessly to establish strong cross-functional and interdisciplinary ties that bind discovery and development into a more cohesive, more effective R&D engine. Much of our success springs from being nimble and pragmatic on the journey: by optimizing areas we know work well and adapting to ever-changing landscapes in the capital markets, therapeutics spaces, and laws and regulations (e.g., IRA).
The Book of Nimbus is still being written, but its arc over the years already shows the shape of what I believe to be Nimbus’ mark on our industry: as an R&D powerhouse with the potential to repeatedly create breakthrough medicines for patients.
1장은 2009년부터 2016년까지 7년간의 이야기입니다. Atlas Ventures의 Bruce Booth와 Schrödinger Ramy Farid 사이의 대화로 시작된 말도 안되는 아이디어 – “2년간 $10 Million을 투자해서 5개의 프로그램을 DC 단계까지 만든다”는 터무니없는 아이디어로 시작합니다. 이 아이디어는 5년에 하나의 DC로 수정되었고 투자금액은 $50 Million으로 올라갔습니다.
ACC Inhibitor인 Firsocostat이 Gilead에 팔렸고 MASH 치료제로 개발 중입니다.
Chapter 1 – An Unreasonable Idea
The year was 2009. Barack Obama had just been sworn in as the 44th president. The automotive industry just received an $81 billion bailout from the federal government, and unemployment sat at 10% (the highest in 25 years). Michael Jackson died, Slumdog Millionaire won the Oscar for Best Picture, and meanwhile Bruce Booth of Atlas Venture and Ramy Farid, CEO of Schrödinger, began work on a very unreasonable idea. It was the beginnings of Project Troubled Water, Inc.: set up a “virtual project team,” leverage Schrödinger scientists to lead computational chemistry, and do all the wet work at CROs. Invest $10 million to get 5 development candidates in 2 years. Unreasonable indeed.
The five development candidates in two years became one development candidate in five years. The cost went from $10 million to $50 million, inclusive of investments in the platform and broader pipeline. In those respects, one might look at Nimbus’ first chapter as a failure, but they’d be wrong. Because the other thing that happened was the creation of an incredible discovery engine that entirely changed the small molecule drug discovery paradigm. Those years of hard work forged the unique project style that coupled computational chemistry leadership with battle-tested medicinal chemists, biologists who are subject matter experts on the target, and CROs and academic collaborators that fueled an unprecedented “DMTA” engine: Design a molecule on a computer, Make the molecule at a CRO, Test the molecule in a proprietary bespoke biological screening cascade for the target, and Analyze the resulting data, which would then feed the design phase of the next iteration. Blood, sweat, and tears poured into the establishment and optimization of this framework. High science led the vanguard of the work, yet behind the scenes a novel business structure was developed simultaneously, the LLC structure. The LLC structure at Nimbus is more than just a holding company framework; it is an intricate, well-planned set of agreements, accounting methods, and governance operations procedures that allowed the Nimbus discovery engine to flourish. Long-time Nimbi extraordinaire Holly Whittemore perfected this approach alongside the amazing counsel at Goodwin, notably Bill Collins, Mitch Bloom, Dan Karelitz, and many others.
By the time Chapter 1 was nearing its end, Project Troubled Water, then Nimbus Discovery, became Nimbus Therapeutics as we took a further step to forward integrate into clinical development. Having partnered our lead IRAK4 asset with Genentech (which ultimately failed to progress), Nimbus entered the clinic with our allosteric inhibitor of acetyl-CoA carboxylase (ACC) in healthy volunteers, with plans to begin a Phase 2 in NASH. Our ACC inhibitor, now named firsocostat, remains first-in-class, and is in a Phase 2b study in severe NASH patients run by Gilead, who acquired the program in 2016.
For further reading about Nimbus’ first chapter, many an excellent blog has been written about those formative days. Check out:
2장은 2016년부터 2022년까지 6년간의 이야기입니다. Fircocostat을 Gilead에 매각한 후 회사의 미래에 대한 논의가 있었고 BD를 통해 2017년 Celgene과 Tyk2와 STING program에 대한 Option deal을 합니다. 그런데 2019년초에 BMS가 Celgene을 합병하면서 Tyk2 프로그램에 위기가 옵니다. BMS는 Nimbus의 Tyk2 program을 back-up으로 가져갔고 BMS가 가진 Tyk2 Inhibitor인 Sotyktu Deucravacitinib) 이 승인이 나면서 Nimbus의 Allosteric Tyk2 product는 Psoriasis 치료제로 선회하게 됩니다.
Phase-3 data package를 가지고 빅파마 들과 딜을 한 결과 Takeda에 매각됩니다.
Chapter 2 – Building an Integrated Approach
It was 2016, we had sold our lead asset to Gilead, and we had no idea what exactly was going to happen next. The transaction in 2016 was also the first true biotech “exit” of a holding company/single asset that would return capital to investors and employees – like a true M&A – but preserve the rest of the portfolio and the Nimbus business model. Miraculously, everyone came back to work the next day, week, month, and it truly felt like a new adventure as we knew we were charting a course not many previously had. The transition had its challenges though: we had begun working in clinical development, hired staff, and now were reset to an early-stage preclinical company. All our resources in chapter 1 had begun funneling to the lead program, and with only $67 million raised over 7 years, Nimbus was not exactly “robust” at an enterprise-level. We had just 22 people by the end of that year, 15% of the company having departed following the Gilead deal.
At that time the Nimbus Board discussed the next chapter of our company. The first thought was to never raise money again; become a perpetual motion machine. We kept 5% of the Gilead deal proceeds in 2016 in the hopes we could span our way to a next BD deal in our pipeline – and we did! In 2017 we formed a classic Celgene option deal with our two most advanced programs, TYK2 and STING. Nimbus retained full ownership and control of the programs in exchange for funding and pre-programmed exits of $400 million each for Phase 1b data in a few years.
That created a conundrum. With the two lead programs effectively pre-sold, what would the rest of Nimbus do? Would we wind down and exit, or chart some different path? That critical strategic discussion led to some fundamental changes in Nimbus, changes that ultimately laid the groundwork for amazing success in chapter 2.
The year was 2018, and the Board at Nimbus had agreed with our plan to re-invest in discovery and build out our internal pipeline. The successful computational powerhouse DMTA cycle we had built could broaden applications across more targets. And in that new pipeline, our goal was to identify “The One” (I personally cannot help but think of Keanu Reeves’ character Neo, from the Matrix movies). “The One” was a molecule that we would forward-integrate further around, which would be the nucleus to crystallize our clinical development organization. The strategic shift spawned our mission statement at Nimbus: We design breakthrough medicines. It also led to a $65 million equity financing to kickstart pipeline creation. Little did we know that “The One” would be a molecule we already had in our hands, our allosteric TYK2 inhibitor….
This direction and change in strategy fomented uncertainty, which led to inevitable turnover. Nine years in at that point, we saw 25% of the Nimbi depart that year, including our first CEO, Don Nicholson, and I am humbled the Board asked me to step in as Nimbus’ next CEO. Having said farewell earlier to our founding CSO, Rosana Kapeller, my first step was to rebuild the fundamental high science foundation of the company. I turned to my good friend and former colleague, Peter Tummino, then VP, Global Head of Lead Discovery at Janssen, to be our next CSO. Over the next four years, the science at Nimbus flourished, and with it, the reputation for excellence grew. We became the magnet for top talent that Nimbus is now known for, attracting such amazing scientists as Christine Loh, Scott Edmondson, Mark Cartwright, and so many more, too many to name, but all of whom deserve my most humble thanks for joining on this mission to design breakthrough medicines for patients.
In the middle of chapter 2, the most dramatic wrinkle then occurred. It was January 3rd, 2019, and BMS just announced that they were acquiring Celgene. I remember learning of this from Holly Whittemore, as I cheerfully greeted her with “Happy New Year” on my first day back to the office. She replied, “Hey, did you see this?” and swiveled her monitor to show me the news. After I picked my lower jaw up off the floor, I said 30 seconds later “We are going to keep our TYK2 program.” Celgene had signed up for the option deal with Nimbus in 2017 to access our (hoped-for at the time) best-in-class allosteric TYK2 inhibitor to compete with their rival BMS’ TYK2 inhibitor (which today is known as Sotyktu). BMS had just begun Phase 3 trials of their agent, which was likely to be successful — as we now know it was.
The initial interactions with BMS were pragmatic and sensible. Following the close of the BMS deal, the Nimbus TYK2 option was allowed to persist as a backup option, should the BMS TYK2 drug fail in Phase 3. During the year-of-Covid, 2020, we slowly started our Phase 1 program with our TYK2 candidate while BMS slogged through Phase 3. Then came 2021, the most consequential, and tumultuous, year in the Book of Nimbus thus far. It was a period of dramatic activity, much of it well-documented in the public domain, but thankfully all resolved by January of 2022. In the end, after a rollercoaster of legal ups and downs, we settled out of court, leaving Nimbus sole ownership of its TYK2 program.
Throughout this period of interacting with BMS, litigation attorneys, and the FTC, Nimbus was fortunate to find investors who believed in our team, our science, and our conviction that we had a sound strategy that did not rely on a binary outcome of whether we won or lost litigation. We were fortunate to raise $225 million to power TYK2, as well as the rest of our pipeline, including the clinical start of our HPK1 inhibitor program in cancer patients. That funding enabled the two Phase 2b trials of our TYK2 program, one in psoriasis and one in psoriatic arthritis.
In 2022, with sole ownership of our lead asset, Nimbus began seriously considering an initial public offering after 13 years of private operation. Our CFO, Ian Sanderson, had joined to lead us through that transition, and instead his experience and expertise led us through finding private funding at the start of a very turbulent period in the public capital markets. By mid-summer 2022, the market sentiment was downright sour, and Nimbus was running low on the cash needed to power up our programs. In true Nimbus fashion, we did continue to keep all our options on the table, and the BD team at Nimbus had been in constant communication since 2019 with all key pharma partners that would entertain talks about our TYK2 program. Our Phase 2b study was wrapping up and we expected data in Q4; meanwhile, on every investor’s mind was the expected approval of BMS’ TYK2 inhibitor in September. Nearly 90% of investors and physicians had predicted that BMS would get a black box warning on Sotyktu, since TYK2 was a JAK family member, even though Sotyktu was super selective against the other JAKs. When the approval finally arrived at 11pm on the PDUFA date with no black box, suddenly, allosteric TYK2s were a new class of medicines for psoriasis with potential in many autoimmune diseases.
Shortly after the Sotyktu approval, our 260 patient Phase 2b study read out. The data, ultimately unveiled at AAD in March 2023, were stunning: biological efficacy rivaling IL-17 and IL-23 with an oral small molecule, and possessing a safety profile at least as benign as BMS’ Sotyktu. Our long-time clinical development lead, Bhaskar Srivastava, an M.D. Ph.D. dermatologist, could not contain his excitement. He delivered one of the most well designed and executed studies in the field and deserves enormous credit for developing a medicine with such profound potential for so many patients.
In the frenetic weeks that followed the unblinding of the study, our Chief Business Officer, Abbas Kazimi, was on center stage, building a team including the excellent advice of Phil Ross at J.P. Morgan and wise counsel of Sarah Solomon at Goodwin. The pharma relationships Nimbus had established allowed diligence teams to engage efficiently and move past the first point of an interaction – trust. Our small team was able to support multiple major pharmaceutical companies plowing through diligence, not just withstanding the onslaught but in fact delivering a data package of Phase 3-ready quality. The bidding was fast and furious, and ultimately the incredible team at Takeda, led by CEO Christoph Weber and R&D Head Andy Plump, became the most compelling group dedicated to taking our program forward to patients, which we announced on December 13, 2022. We could not be more thrilled with their leadership and commitment, and we closed the deal by February of 2023.
For further reading about Nimbus’ second chapter, many excellent blogs were written about this period. Check out:
2023년 이후 Nimbus는 3장을 쓰기 시작했습니다. 새로운 Breakthrough Medicine을 개발한다는 계획입니다. 구조조정이 있었고 Oncology와 Immunology Programs를 개발하고 있습니다.
Chapter 3 – Establishing a Legacy R&D Institution
This blog is being written as we turn the page to chapter 3, however the groundwork began with long-range planning almost a year ago. We had scenarios for every eventuality for the TYK2 data, partnering interest, and the financing environment. With that said, we knew if we were successful in psoriasis, the implications would require a large multinational company to create the value of global registrations in multiple indications. Given the value of established infrastructure in pharma, it was clear that an M&A acquisition of our TYK2 subsidiary was likely.
We therefore have had some time in which to contemplate what this next act for Nimbus holds. Although we are just now at the beginning stages of the great journey to come over the coming years, many of the formative pieces are now in place — just as our TYK2 program was at the time of Nimbus’ last inflection point. Our clinical-stage HPK1 inhibitor is now progressing into expansion trials in solid tumors, while a crafted pipeline of opportunities, including what we would consider a disruptive medicine in the autoimmune field, heads toward the clinic next year. While our expertise in immunology and oncology is strong, we also have depth in metabolic disorders, and have a fabulous collaboration with Eli Lilly on AMPK activators, along with earlier programs in discovery.
And excitingly, we are better positioned than at any point in our history to navigate what comes next. Our investments throughout chapter 2 have built an organization with an even wider skillset, from discovery through to clinical execution, and deeper disease area expertise than ever before. Key to Nimbus’ third chapter will be Chief Medical Officer Nathalie Franchimont, who joined us from Biogen late last year to lead our Development organization, building upon our foundations of quality, operations, and execution. Nathalie, Peter Tummino and Bhaskar Srivastava are building out our early clinical and translational biology expertise, while at the same time we are investing in our computational capabilities, tackling tough targets like transmembrane GPCRs in our discovery pipeline. As the winds of change in our industry keep blowing strong, the flexibility and optionality of the Nimbus structure remain a key competitive advantage that has contributed to this enterprise’s longevity.
Transitions are not easy times, though, and as was the case in our move to chapter 2, we’ve needed to navigate turnover and figure out a way to realign and reorganize the Nimbi while preserving our diversity and special sauce, a task that has been led with care and grace by our Chief People Officer, Erin Cowhig. Reorganizations are never a pleasant task, and it has led to some tough choices, where we have needed to bid farewell to some excellent Nimbi simply because their roles were not going to be essential to this next chapter. We thank them for their service and are committed to their safe landings, as they join the ranks of amazingly successful alumni from Nimbus. We’re proud of the small but growing Nimbus diaspora, a testament to how special a place Nimbus has become. Elsewhere in the industry we see biotechs who emulate our corporate structure, our computational engine, or our approach to deal-making (or all 3!). Awesome. We must be doing something right. If Nimbus is able to help shape the industry approach, give a better shot to making therapeutics that help patients, then we have multiplied our impact many times beyond our four walls.
Nimbus is committed to the notion that “small is beautiful” in drug R&D: breakthrough small molecules designed by a small expert team. We have built hard-earned capabilities in both discovery and development, and will continue to build on those in chapter 3. Our mission remains the same: We design breakthrough medicines. Our objective in dollars and cents terms is to again shoot for the moon, to become again a multi-billion-dollar biotech. But our ambition is far greater than that. Nimbus has an opportunity to build a legacy R&D institution. A paradigm of excellence in small molecule drug discovery and development. Chapter 3 will take some time to evolve as the pages are just being written, but we are blessed with an ideal combination of functional skills, established quality processes, and enough hungry, “unreasonable” individuals who drive us to become more than we ever thought we could be.
Nimbus의 14-15년간의 실험의 경험을 가지고 IFM Therapeutics가 2017년에 Series A를 통해 Stealth-mode로 부터 알려진 이후에 지금까지 IFM Uno라는 자회사를 BMS에 매각하면서 STING Agonist와 NLRP3 Agonist를 넘겼고 2021년에는 Novartis에 IFM TRE를 통해서 NLRP3 Antagonist program을 팔았습니다. 그리고 오늘 다시 IFM Due를 통해서 Novartis는 STING Antagonist program을 매입했습니다.
Atlas의 Option-to-buy M&A를 위한 스타트업 설립은 최근 뜸해졌지만 오랜 기간의 경험이 축적된 만큼 현재 바이오텍 스타트업계에서 이런 구조를 가장 잘 활용하고 운용하는 벤처캐피탈이 아닐까 생각합니다.
Today IFM Therapeutics announced the acquisition of IFM Due, one of its subsidiaries, by Novartis. Back in Sept 2019, IFM granted Novartis the right to acquire IFM Due as part of an “option to buy” collaboration around cGAS-STING antagonists for autoimmune disease.
This secures for IFM what is a rarity for a single biotech company: a liquidity hat trick, as this milestone represents the third successful exit of an IFM Therapeutics subsidiary since its inception in 2015.
Back in 2017, BMS purchased IFM’s NLRP3 and STING agonists for cancer. In early 2019, Novartis acquired IFM Tre for NLRP3 antagonists for autoimmune disease, which are now being studied in multiple Phase 2 studies. Then, later in 2019, Novartis secured the right to acquire IFM Due after their lead program entered clinical development. Since inception, across the three exits, IFM has secured over $700M in upfront cash payments and north of $3B in biobucks.
Kudos to the team, led by CEO Martin Seidel since 2019, for their impressive and continued R&D and BD success.
Option-to-Acquire Deals
These days option-based M&A deals aren’t in vogue: in large part because capital generally remains abundant despite the contraction, and there’s still a focus on “going big” for most startup companies. That said, lean capital efficiency around asset-centric product development with a partner can still drive great returns. In different settings or stages of the market cycle, different deal configurations can make sense.
During the pandemic boom, when the world was awash in capital chasing deals, “going long” as independent company was an easy choice for most teams. But in tighter markets, taking painful levels of equity dilution may be less compelling than securing a lucrative option-based M&A deal.
For historical context, these option-based M&A deals were largely borne out of necessity in far more challenging capital markets (2010-2012) on the venture front, when both the paucity of private financing and the tepid exit environment for early stage deals posed real risks to biotech investment theses. Pharma was willing to engage on early clinical or even preclinical assets with these risk-sharing structures as a way to secure optionality for their emerging pipelines.
As a comparison, in 2012, total venture capital funding into biotech was less than quarter of what it is now, even post bubble contraction, and back then we had witnessed only a couple dozen IPOs in the prior 3 years combined. And most of those IPOs were later stage assets in 2010-2012. Times were tough for biotech venture capital. Option-based deals and capital efficient business models were part of ecosystem’s need for experimentation and external R&D innovation.
Many flavors of these option-based deals continued to get done for the rest of the decade, and indeed some are still getting done, albeit at a much less frequent cadence. Today, the availability of capital on the supply side, and the reduced appetite for preclinical or early stage acquisitions on the demand side, have limited the role of these option to buy transactions in the current ecosystem.
But if the circumstances are right, these deals can still make some sense: a constructive combination of corporate strategy, funding needs, risk mitigation, and collaborative expertise must come together. In fact, Arkuda Therapeutics, one of our neuroscience companies, just announced a new option deal with Janssen.
Stepping back, it’ s worth asking what has been the industry’s success rate with these “option to buy” deals.
Positive anecdotes of acquisition options being exercised over the past few years are easy to find. We’ve seen Takeda exercise its right to acquire Maverick for T-cell engagers and GammaDelta for its cellular immunotherapy, among other deals. AbbVie recently did the same with Mitokinin for a Parkinson’s drug. On the negative side, in a high profile story last month, Gilead bailed on purchasing Tizona after securing that expensive $300M option a few years ago.
But these are indeed just a few anecdotes; what about data since these deal structures emerged circa 2010? Unfortunately, as these are mostly private deals with undisclosed terms, often small enough to be less material to the large Pharma buyer, there’s really no great source of comprehensive data on the subject. But a reasonable guess is that the proportion of these deals where the acquisition right is exercised is likely 30%.
This estimate comes from triangulating from a few sources. A quick and dirty dataset from DealForma, courtesy of Tim Opler at Stifel, suggests 30% or so for deals 2010-2020. Talking to lawyers from Goodwin and Cooley, they also suggest ballpark of 30-50% in their experience. The shareholder representatives at SRS Acquiom (who manage post-M&A milestones and escrows) also shared with me that about 33%+ of the option deals they tracked had converted positively to an acquisition. As you might expect, this number is not that different than milestone payouts after an outright acquisition, or future payments in licensing deals. R&D failure rates and aggregate PoS will frequently dictate that within a few years, only a third of programs will remain alive and well.
Atlas’ experience with Option-based M&A deals
Looking back, we’ve done nearly a dozen of these option-to-buy deals since 2010. These took many flavors, from strategic venture co-creation where the option was granted at inception (e.g., built-to-buy deals like Arteaus and Annovation) to other deals where the option was sold as part of BD transaction for a maturing company (e.g., Lysosomal Therapeutics for GBA-PD).
Our hit rate with the initial option holder has been about 40%; these are cases where the initial Pharma that bought the option moves ahead and exercises that right to purchase the company. Most of these initial deals were done around pre- or peri-clinical stage assets. But equally interesting, if not more so, is that in situations where the option expired without being exercised, but the asset continued forward into development, all of these were subsequently acquired by other Pharma buyers – and all eight of these investments generated positive returns for Atlas funds. For example, Rodin and Ataxion had option deals with Biogen that weren’t exercised, and went on to be acquired by Alkermes and Novartis. And Nimbus Lakshmi for TYK2 was originally an option deal with Celgene, and went on to be purchased by Takeda.
For the two that weren’t acquired via the option or later, science was the driving factor. Spero was originally an LLC holding company model, and Roche had a right to purchase a subsidiary with a quorum-sensing antibacterial program (MvfR). And Quartet had a non-opioid pain program where Merck had acquired an option. Both of these latter programs were terminated for failing to advance in R&D.
Option deals are often criticized for “capping the upside” or creating “captive companies” – and there’s certainly some truth to that. These deals are structured, typically with pre-specified return curves, so there is a dollar value that one is locked into and the presence of the option right typically precludes a frothy IPO scenario. But in aggregate across milestones and royalties, these deals can still secure significant “Top 1%” venture upside though if negotiated properly and when the asset reaches the market: for example, based only on public disclosures, Arteaus generated north of $300M in payments across the upfront, milestones, and royalties, after spending less than $18M in equity capital. The key is to make sure the right-side of the return tail are included in the deal configuration – so if the drug progresses to the market, everyone wins.
Importantly, once in place, these deals largely protect both the founders and early stage investors from further equity dilution. While management teams that are getting reloaded with new stock with every financing may be indifferent to dilution, existing shareholders (founders and investors alike) often aren’t – so they may find these deals, when negotiated favorably, to be attractive relative to the alternative of being washed out of the cap table. This is obviously less of a risk in a world where the cost of capital is low and funding widely available.
These deal structures also have some other meaningful benefits worth considering though: they reduce financing risk in challenging equity capital markets, as the buyer often funds the entity with an option payment through the M&A trigger event, and they reduce exit risk, as they have a pre-specified path to realizing liquidity. Further, the idea that the assets are “tainted” if the buyer walks hasn’t been borne out in our experience, where all of the entities with active assets after the original option deal expired were subsequently acquired by other players, as noted above.
In addition, an outright sale often puts our prized programs in the hands of large and plodding bureaucracies before they’ve been brought to patients or later points in development. This can obviously frustrate development progress. For many capable teams, keeping the asset in their stewardship even while being “captive”, so they can move it quickly down the R&D path themselves, is an appealing alternative to an outright sale – especially if there’s greater appreciation of value with that option point.
Option-based M&A deals aren’t right for every company or every situation, and in recent years have been used only sparingly across the sector. They obviously only work in practice for private companies, often as alternative to larger dilutive financings on the road to an IPO. But for asset-centric stories with clear development paths and known capital requirements, they can still be a useful tool in the BD toolbox – and can generate attractive venture-like returns for shareholders.
Like others in the biotech ecosystem, Atlas hasn’t done many of these deals in recent funds. And it’s unlikely these deals will come back in vogue with what appears to be 2024’s more constructive fundraising environment (one that’s willing to fund early stage stories), but if things get tighter or Pharma re-engages earlier in the asset continuum, these could return to being important BD tools. It will be interesting to see what role they may play in the broader external R&D landscape over the next few years.
Most importantly, circling back to point of the blog, kudos to the team at IFM and our partners at Novartis!
IFM Therapeutics, a biopharmaceutical company developing a portfolio of first-in-class small molecules targeting the innate immune system, today announced the closing of a $27 million Series A financing led by Atlas Venture and Abingworth, with participation from Novartis. In conjunction with the funding Jean-François Formela and Vincent Miles, Partners at Atlas and Abingworth respectively, have joined IFM’s CEO, Gary D. Glick, on the board of directors, with Dr. Formela serving as chair of the board.
IFM Therapeutics, incubated as a part of the Atlas Venture seed program, is developing modulators of novel targets that either enhance innate immune responses for treating cancer, or dampen certain immune responses that drive many inflammatory diseases. The company will use the proceeds of the financing to advance and expand its early-stage portfolio and begin clinical development of its most advanced product candidate, a selective activator of a novel target, for treating solid tumors.
“While proteins in the innate immune system represent an attractive landscape of therapeutic targets, they have been notoriously difficult to drug,” said Dr. Formela from Atlas. “During the brief period since its founding, IFM has made excellent progress on several of these targets, reflecting its exceptional team of experienced scientists and executives, possessing expertise in medicinal chemistry, a deep understanding of the relevant biology, and relationships with academic thought leaders in the areas of immunology and immune oncology.”
“IFM’s programs have the potential to make a major difference in the lives of patients with serious, and sometimes fatal, diseases,” said Abingworth’s Dr. Miles. “We look forward to helping the team build on their strong start to advance these programs into clinical development.”
“This financing is an important validation of the IFM team and technology,” said Dr. Glick, IFM’s Co-founder and CEO. “The company is fortunate to be working with a talented and experienced group of investors. Their expertise in building world-class biopharmaceutical companies will be invaluable as we grow the company.”
살다보면 진짜와 가짜를 구분하는 눈이 생기는 것 같습니다. 뜬금없이 무슨 말이냐? 고 할 수 있겠지만 어떤 사람은 말은 번듯이 해도 자신이 경험하지 않은 것을 얘기해서 혼란을 주기도 하고 어떤 사람은 투박하지만 진짜로 자신이 경험한 것을 말하는 사람이 있습니다.
편정현 헤드헌터는 후자에 속하는 사람이라고 생각합니다. 나이가 들어서 정년퇴직을 당하는 (?) 경우가 많은데 이에 대해 편정현 헤드헌터는 절대 좌절하게 하지도 않고 그렇다고 해서 돈을 굴리라는 둥 뜬구름 잡는 말을 하지도 않습니다. 그대신 퇴직금을 3년에 나눠서 생활비로 쓰면서 공부를 해서 3년후부터는 벌 생각을 하라고 말합니다.
아래의 유튜브 영상은 편정현님의 자기 경험담입니다. 대리운전을 3년여를 하면서 배운 것에 대해 말씀하시고 계십니다.
경기불황 속 인재전쟁이 다시 시작된다. 뛰어난 인재를 채용하기 더 어려워지고 있다. 최근 뉴스에서는 우리나라가 경기불황에 접어들고 있으며 이 상황이 장기간 지속될 것이라는 전망을 내놓고 있다. 경기불황은 인재전쟁 시대의 서막이다. 회사 입장에서는 경제 상황이 어려워질수록 뛰어난 인재를 확보하기 어려워진다.
인재를 확보하기 위한 보편적인 방법 중 하나는 헤드헌터다. 헤드헌터는 문자 그대로 풀이하면 ‘머리를 사냥하는 사람’이다. 단어 자체의 느낌은 살벌하지만, 머리처럼 중요한 인재를 확보할 수 있다는 점에서 적절한 용어라고 할 수 있다. 헤드헌팅은 대공황을 겪었던 1930년 미국에서 새로운 채용방식으로 등장해, 현재까지 경력인재에 대한 수요를 충족시키는 방법으로 활용되는 중이다. 컨설팅 업체 ‘인파트너스’의 편정현 대표 헤드헌터가 인재전쟁에서 승리할 수 있는 노하우에 대해 밝혔다. PD 출신으로 방송계에서만 15년 이상의 경력을 쌓은 베테랑인 편정현 씨는 삼성, SK, 한화, 하나금융그룹 등과 같이 굵직한 대기업에서의 채용 프로젝트를 연달아 성공시킨 경험이 있다. 대기업뿐만이 아니다 인재 한 명 한 명이 소중한 스타트업도 많이 활용을 하고 있다.
-인재를 확보하기 위해 헤드헌터로서 갖춰야 할 역량은 무엇인가? “우선, 소명의식이 있어야 한다. 헤드헌터는 인재전쟁에서 고객사를 대신해 싸우는 용병이다. 용병의 가장 큰 문제는 충성심의 결여다. 훌륭한 용병이 되기 위해서는 용병으로서의 소명의식을 갖춰야 한다. 스위스 용병이 아직도 교황청을 지키는 이유는 500년 전 200여 명의 근위병 중 150명이 목숨을 잃으면서까지 교황을 지켰기 때문이다. 헤드헌터도 마찬가지다. 돈에 의해서만 움직이는 헤드헌터는 좋은 헤드헌터라고 할 수 없다. 소명의식이 있는 헤드헌터가 되어야 한다.
또한, 고객의 요구사항과 지시사항을 명확하게 파악하기 위해 끊임없이 소통해야 한다. 고객의 언어와 고객의 눈높이에 맞춰 소통하는 능력은 헤드헌터의 기본이다. 헤드헌터의 고객인 고객사와 후보자의 이야기를 경청하는 자세가 바탕이 돼야 한다. 여기에 자신만의 도구가 있어야 한다. 보통 헤드헌터들은 다양한 툴을 활용한다. 여러 툴을 활용해 상황에 따라 그물을 넓게 또는 좁게 던지며 빠르게 인재를 선별해야 한다. 그리고 적절한 인재를 적절한 포지션에 추천하는 것도 중요하다. 나도 여러 인재 서치 툴 및 데이터베이스나 플랫폼을 사용하는 중이다.
특히 ‘위크루트’라는 플랫폼을 애용한다. ‘위크루트’는 기업 인사담당자와 헤드헌터를 연결해주는 헤드헌팅 플랫폼이다. 위크루트의 조강민 대표는 10년 이상 대기업 인사담당자로 일하면서 인재전쟁에서 승리하는 방법을 끊임없이 고민해왔다.”
-인재전쟁에서 승리하기 위한 방법은 무엇인가? “뛰어난 인재를 확보하기 위해서는 뛰어난 헤드헌터를 확보해야 한다. 결국 가장 헤드헌터가 많은 곳이 가장 사람을 잘 찾지 않겠는가? ‘위크루트’는 서비스 출시 1년 만에 430명 이상의 헤드헌터가 가입해 활동하는 국내 최대 헤드헌팅 네트워크로 자리매김했다. 기업 입장에서는 수백 명의 헤드헌터가 필요한 인재를 찾아주는 플랫폼을 이용하는 것도 하나의 방법이다.”
-헤드헌팅 플랫폼 ‘위크루트’만의 특징은 무엇인가? “근성 있는 헤드헌터가 모여있는 곳이다. 인원 수로도 이미 국내 최고지만 헤드헌터의 역량도 최고라고 평가할 수 있다. 헤드헌팅 프로젝트가 주어지면 어떻게든 성공시키는 헤드헌터들이 모여있다. ‘위크루트’를 이용하는 헤드헌터에 대한 믿음이 있다. 또한, 가장 혁신적인 플랫폼이다. ‘위크루트’는 헤드헌팅 플랫폼으로는 최초로 3개의 특허가 등록되었거나 등록 과정에 있다. 아이디어가 사업화되는 일련의 과정을 혁신이라고 부른다는 점에서, ‘위크루트’는 가장 혁신적인 플랫폼이다. 한 번 사용한 인사담당자들의 재사용률은 90% 이상이다.
여기에 소통이 편리한 플랫폼이다. 기존의 인사담당자와 헤드헌터 간 이메일을 통한 커뮤니케이션의 한계를 보완해준다. 특히 소통 방식의 개선으로 한화그룹 채용담당자의 업무 효율이 300% 이상 올라갔다고 한다. 삼성, SK, KT와 같은 대기업 인사담당자가 사용하는 이유 중 하나다.
06 July 2025 Update
편정현님에 대한 보다 업데이트된 기사가 있어서 글을 올립니다. 편정현님의 좋은 조언을 보면서 저 또한 베이비부머 세대로서 좀더 고민하고 새로운 기술에 도전해 봐야겠다고 생각했습니다. 아래는 기사입니다.
미국과 일본에서 연구비를 받아서 초기의 연구를 진행하다가 2021년에 $37 Million Series A를 했습니다. 현재 Self-Amplifying RNA-LNP 백신인 COVID-19 Vaccine이 임상3상을 진행 중이고 VLP 백신인 Malaria 백신이 임상2상에 있고 VLP 백신인 Dengue Vaccine이 IND-enabling study를 진행하고 있습니다.
2017년에 미국 NIH SBIR grant를 Cancer vaccine 연구과제로 받았습니다.
Cancer immunotherapy has revolutionized today s treatments for many forms of cancers In particular blocking antibodies to immune checkpoint proteins such as programmed cell death PD and its ligand programmed cell death ligand PD L effectively unleash immune cell destruction of cancerous cells. However the high cost of these antibody-based immunotherapies their intensive therapeutic regimen and availability only at specialized medical centers make current immunotherapies impractical in all but the most advantaged societies. We hypothesize that an effective vaccine which can overcome these significant shortcomings of antibody-based immunotherapies while simultaneously mimicking their potent therapeutic benefits can impact the lives of countless more patients in particular a vaccine that induces durable levels of host antibodies against the tumor associated PD L protein will have powerful anti tumor effects by inhibiting the PD L PD immunosuppressive checkpoint interaction. We at VLP Therapeutics have developed a proprietary plug and play vaccine platform called inserted alphavirus virus like particle i VLP using the Chikungunya CHIK VLP VLPs mimic the conformation of native viruses without the viral genome thus capable of stimulating a robust host response absent safety issues Foreign antigens can be inserted into the surface loop domains of i VLP Due to its unique structure i VLP can efficiently present a dense array of copies of the inserted antigen on the particle surface i VLP induces highly effective immune responses to the inserted antigen and CHIK VLPs have shown acceptable safety profiles in a Phase I clinical trial. We propose to establish proof of concept of our PD L targeting vaccine s efficacy in a triple negative breast cancer TNBC like model given that TNBCs have shown some responsiveness to PD L PD antibody therapies in clinical trials and there remains a high unmet need for effective therapies against this particularly aggressive form of breast cancer which has the worst five year survival prognosis among all breast cancers and for which standard chemotherapy treatment is largely ineffective. The goal of this proposal is to determine the extent to which vaccination with our proprietary i VLP based PD L vaccine PD L VLP can mimic the effects of passive PD L monoclonal antibody therapy This study will provide the basis for advancing this exciting approach into clinical trials To create this vaccine we propose the following Specific Aims:
Aim: Determine the ability of PD L VLP to stimulate the production of antibodies that bind to tumor associated PD L and inhibit its binding to the T cell immune checkpoint receptor PD in murine models.
Aim: Determine the therapeutic effects of PD L VLP vaccination in a PD PD L antibody therapy sensitive murine tumor model benchmarked against PD L monoclonal antibody treatment.
Aim: Determine whether potential immune related adverse events including autoimmunity can be induced by our PD L VLP vaccine Narrative Cancer immunotherapy in particular blocking antibodies to immune checkpoint proteins effectively unleash immune cell destruction of cancerous cells However their high cost intensive therapeutic regimen and limited availability only at sophisticated medical centers make current antibody based immunotherapies impractical in all but privileged societies An effective vaccine directed against the tumor associated PD L protein which mimics the therapeutic benefits of antibody based PD L immunotherapy while overcoming its significant shortcomings described above will have powerful anti tumor effects.
3월의 시리즈 A와 12월의 시리즈 A-1에 모두 SK Impact Fund가 참여를 했습니다. 코로나 팬데믹 동안에 COVID-19 Vaccine 개발로 펀딩이 될 수 있었던 것 같습니다. 일본 바이오텍 벤처는 미국과 달리 서서히 기술개발을 이어가면서 공동연구를 주로 하는 방법으로 회사가 성장을 합니다. VLP Therapeutics는 saRNA-LNP 혹은 saRNA-VLP 백신 개발을 하는데 기대가 많이 됩니다.
US-based biotech company VLP Therapeutics, Inc. (VLPT) announced on March 15 that it has raised US$16 million in a Series A funding round from MIYAKO Capital Co., Ltd., Sojitz Corporation, Konishiyasu Co., Ltd. in Japan, and three existing investors in the US (Mr. Robert G. Hisaoka, SK Impact Fund, LLC, and RJ Fund, LLC) for research and development of a cancer treatment vaccine. With this funding, VLPT aims to accelerate the project well underway in the US and move into clinical trials at the earliest date possible.
“I have a high hope for VLP Therapeutics to become a leading company with its innovative platform technologies in the fields of infectious diseases and cancer, deep-rooted with CEO Wataru Akahata’s continuing commitment to virus and vaccine research and extensive experience of clinical trials,” says Dr. Hiroyuki Misawa, director and partner of MIYAKO Capital Co., Ltd. “It is my great pleasure to back up, as a shareholder, a biotech company with such advanced technologies.”
“I am optimistic that the innovative vaccines now being developed at VLP Therapeutics will make a significant contribution to the treatment of cancer, the prevention of malaria and dengue fever, and the fight against new threats such as COVID-19. In turn, I believe this will improve health and well-being for all and advance the development of medicine,” says Masayoshi Fujimoto, president and CEO of Sojitz Corporation. “We, Sojitz Corporation, are very pleased to work with VLPT CEO Wataru Akahata, an ambitious scientist with considerable experience in vaccine R&D, as well as with the members of the research team and the company founders who are well-versed in pharmaceutical development. We will do whatever we can to help VLPT grow going forward.”
“Since its inception over 190 years ago, we, Koshishiyasu, have been devoted to making the world a better place through the sales of industrial chemicals. It is therefore our great honor to invest in the cancer treatment vaccine R&D underway at VLP Therapeutics, which is also combating malaria and Covid-19 with its novel technologies.” says Toshiyuki Konishi, president and CEO of Konishiyasu Co., Ltd. “We are confident that, by financially supporting VLPT, we can contribute to the well-being of society, and they will make further progress in their vaccine R&D efforts.”
US-based biotech company VLP Therapeutics, Inc. (VLPT) announced on December 27 that it has signed an agreement for an investment of US$21 million in a Series A-1 round from six investors, consisting of two new investors: Nobelpharma Co., Ltd. and MUFG Bank, Ltd., and four existing investors: Sojitz Corporation, MIYAKO Capital Co., Ltd., Mr. Robert G. Hisaoka and SK Impact Fund, LLC.
With this funding, VLPT aims to further accelerate the research and development of a cancer treatment vaccine as well as prophylactic vaccines against malaria, dengue, etc. and move into clinical trials at the earliest date possible. This is an additional investment following US$16 million raised in a Series A round in March 2021.
“I have long committed to the research and development of vaccines against cancer and infectious diseases so the people across the globe can lead normal lives,” says Wataru Akahata, CEO and co-founder of VLPT. “We were fortunate enough to be able to raise funding in March to facilitate our cancer treatment vaccine R&D. This additional funding will now allow us to further accelerate our other R&D efforts in infectious diseases area as well. This means a lot as it helps us to push our scientific endeavors forward at much faster pace, enabling us to get one step closer in making a greater social impact.”
About VLP Therapeutics: VLP Therapeutics, Inc. (VLPT), co-founded in 2013 by Drs. Wataru Akahata, Ryuji Ueno, and Sachiko Kuno, is a Gaithersburg, MD-based biotech company with a mission to address unmet medical needs worldwide and expand the frontiers of vaccine treatment. Led by CEO Akahata VLPT is currently engaged in research and development of a cancer treatment vaccine as well as prophylactic vaccines against malaria, dengue, etc. using VLPT’s proprietary platform technologies.
Cell Reports에 낸 논문에 saRNA-LNP COVID-19 백신의 임상1상 결과를 보고했는데 BioNTech의 백신 30 mcg과 비교하여 VLP의 3 mcg 용량으로도 동등하거나 우수함을 증명했습니다. 기존 백신과 차별점은 RBD-anchoring을 한 것이 특징입니다.
저는 Science Writer나 Medical Writer에 대한 생각이 있는데요. 이 분야에서 앞서가는 한 분을 저는 흠모하고 있습니다.
Elie Dolgin 박사는 University of Edinburgh에서 진화유전학 (Evolutionary Genetics)으로 박사학위를 받고 Science Journalist로 활동을 하는 분입니다.
STAT, Nature Medicine 이나 The Scientist 에 글을 올리고 있고 . The New York Times, Science, IEEE Spectrum, Nature 와 같은 저널에 글을 기고하는 Freelancer Science Journalist입니다. 저는 Elie Dolgin 박사의 글의 정교한 스타일을 좋아하는데요. 정말 오랜 기간 인터뷰와 준비를 거쳐서 글이 다음어진 것이 느껴집니다.
대표적인 글로 2021년에 쓴 아래의 글이 있습니다. mRNA Vaccine이 나오기까지의 과학자들의 노력에 대해 조사하고 글을 쓴 것입니다.
Hundreds of scientists had worked on mRNA vaccines for decades before the coronavirus pandemic brought a breakthrough.
In late 1987, Robert Malone performed a landmark experiment. He mixed strands of messenger RNA with droplets of fat, to create a kind of molecular stew. Human cells bathed in this genetic gumbo absorbed the mRNA, and began producing proteins from it1.
Realizing that this discovery might have far-reaching potential in medicine, Malone, a graduate student at the Salk Institute for Biological Studies in La Jolla, California, later jotted down some notes, which he signed and dated. If cells could create proteins from mRNA delivered into them, he wrote on 11 January 1988, it might be possible to treat RNA as a drug”. Another member of the Salk lab signed the notes, too, for posterity. Later that year, Malone’s experiments showed that frog embryos absorbed such mRNA2. It was the first time anyone had used fatty droplets to ease mRNA’s passage into a living organism.
Those experiments were a stepping stone towards two of the most important and profitable vaccines in history: the mRNA-based COVID-19 vaccines given to hundreds of millions of people around the world. Global sales of these are expected to top US$50 billion in 2021 alone.
But the path to success was not direct. For many years after Malone’s experiments, which themselves had drawn on the work of other researchers, mRNA was seen as too unstable and expensive to be used as a drug or a vaccine. Dozens of academic labs and companies worked on the idea, struggling with finding the right formula of fats and nucleic acids—the building blocks of mRNA vaccines.
Today’s mRNA jabs have innovations that were invented years after Malone’s time in the lab, including chemically modified RNA and different types of fat bubble to ferry them into cells. Still, Malone, who calls himself the“inventor of mRNA vaccines”, thinks his work hasn’t been given enough credit.“ I’ve been written out of history,” he told Nature.
The debate over who deserves credit for pioneering the technology is heating up as awards start rolling out—and the speculation is getting more intense in advance of the Nobel prize announcements next month. But formal prizes restricted to only a few scientists will fail to recognize the many contributors to mRNA’s medical development. In reality, the path to mRNA vaccines drew on the work of hundreds of researchers over more than 30 years.
The story illuminates the way that many scientific discoveries become life-changing innovations: with decades of dead ends, rejections and battles over potential profits, but also generosity, curiosity and dogged persistence against scepticism and doubt.“ It’s a long series of steps,” says Paul Krieg, a developmental biologist at the University of Arizona in Tucson, who made his own contribution in the mid-1980s,“and you never know what’s going to be useful”.
The beginnings of mRNA
Malone’s experiments didn’t come out of the blue. As far back as 1978, scientists had used fatty membrane structures called liposomes to transport mRNA into mouse3 and human4 cells to induce protein expression. The liposomes packaged and protected the mRNA and then fused with cell membranes to deliver the genetic material into cells. These experiments themselves built on years of work with liposomes and with mRNA; both were discovered in the 1960s.
The history of mRNA vaccines: A timeline that shows the key scientific innovations in the development of mRNA vaccines.
Back then, however, few researchers were thinking about mRNA as a medical product—not least because there was not yet a way to manufacture the genetic material in a laboratory. Instead, they hoped to use it to interrogate basic molecular processes. Most scientists repurposed mRNA from rabbit blood, cultured mouse cells or some other animal source.
That changed in 1984, when Krieg and other members of a team led by developmental biologist Douglas Melton and molecular biologists Tom Maniatis and Michael Green at Harvard University in Cambridge, Massachusetts, used an RNA-synthesis enzyme (taken from a virus) and other tools to produce biologically active mRNA in the lab5—a method that, at its core, remains in use today. Krieg then injected the lab-made mRNA into frog eggs, and showed that it worked just like the real thing6.
Both Melton and Krieg say they saw synthetic mRNA mainly as a research tool for studying gene function and activity. In 1987, after Melton found that the mRNA could be used both to activate and to prevent protein production, he helped to form a company called Oligogen (later renamed Gilead Sciences in Foster City, California) to explore ways to use synthetic RNA to block the expression of target genes—with an eye to treating disease. Vaccines weren’t on the mind of anyone in his lab, or their collaborators.
Diptych of portraits of Paul Krieg and Douglas Melton
“RNA in general had a reputation for unbelievable instability,” says Krieg.“ Everything around RNA was cloaked in caution.” That might explain why Harvard’s technology-development office elected not to patent the group’s RNA-synthesis approach. Instead, the Harvard researchers simply gave their reagents to Promega Corporation, a lab-supplies company in Madison, Wisconsin, which made the RNA-synthesis tools available to researchers. They received modest royalties and a case of Veuve Clicquot Champagne in return.
Patent disputes
Years later, Malone followed the Harvard team’s tactics to synthesize mRNA for his experiments. But he added a new kind of liposome, one that carried a positive charge, which enhanced the material’s ability to engage with the negatively charged backbone of mRNA. These liposomes were developed by Philip Felgner, a biochemist who now leads the Vaccine Research and Development Center at the University of California, Irvine.
Despite his success using the liposomes to deliver mRNA into human cells and frog embryos, Malone never earned a PhD. He fell out with his supervisor, Salk gene-therapy researcher Inder Verma and, in 1989, left graduate studies early to work for Felgner at Vical, a recently formed start-up in San Diego, California. There, they and collaborators at the University of Wisconsin–Madison showed that the lipid–mRNA complexes could spur protein production in mice7. (Malone and his Vical coworkers also explored using mRNA for vaccines: their early patent filings describe injecting mRNA coding for HIV proteins into mice, and observing some protection against infection, although not the production of specific immune cells or molecules; this work was never published in a peer-reviewed journal).
Then things got messy. Both Vical (with the University of Wisconsin) and the Salk began filing for patents in March 1989. But the Salk soon abandoned its patent claim, and in 1990, Verma joined Vical’s advisory board.
Malone contends that Verma and Vical struck a back-room deal so that the relevant intellectual property went to Vical. Malone was listed as one inventor among several, but he no longer stood to profit personally from subsequent licensing deals, as he would have from any Salk-issued patents. Malone’s conclusion:“ They got rich on the products of my mind.”
Verma and Felgner categorically deny Malone’s charges.“ It’s complete nonsense,” Verma told Nature. The decision to drop the patent application rested with the Salk’s technology-transfer office, he says. (Verma resigned from the Salk in 2018, following allegations of sexual harassment, which he continues to deny.)
Malone left Vical in August 1989, citing disagreements with Felgner over“scientific judgment”and“credit for my intellectual contributions”. He completed medical school and did a year of clinical training before working in academia, where he tried to continue research on mRNA vaccines but struggled to secure funding. (In 1996, for example, he unsuccessfully applied to a California state research agency for money to develop a mRNA vaccine to combat seasonal coronavirus infections.) Malone focused on DNA vaccines and delivery technologies instead.
In 2001, he moved into commercial work and consulting. And in the past few months, he has started publicly attacking the safety of the mRNA vaccines that his research helped to enable. Malone says, for instance, that proteins produced by vaccines can damage the body’s cells and that the risks of vaccination outweigh the benefits for children and young adults—claims that other scientists and health officials have repeatedly refuted.
Manufacturing challenges
In 1991, Vical entered into a multimillion-dollar research collaboration and licensing pact with US firm Merck, one of the world’s largest vaccine developers. Merck scientists evaluated the mRNA technology in mice with the aim of creating an influenza vaccine, but then abandoned that approach.“ The cost and feasibility of manufacturing just gave us pause,” says Jeffrey Ulmer, a former Merck scientist who now consults with companies on vaccine-research issues.
Researchers at a small biotech firm in Strasbourg, France, called Transgène, felt the same way. There, in 1993, a team led by Pierre Meulien, working with industrial and academic partners, was the first to show that an mRNA in a liposome could elicit a specific antiviral immune response in mice8. (Another exciting advance had come in 1992, when scientists at the Scripps Research Institute in La Jolla used mRNA to replace a deficient protein in rats, to treat a metabolic disorder9. But it would take almost two decades before independent labs reported similar success.)
The Transgène researchers patented their invention, and continued to work on mRNA vaccines. But Meulien, who is now head of the Innovative Medicines Initiative, a public–private enterprise based in Brussels, estimated that he needed at least€100 million (US$119 million) to optimize the platform—and he wasn’t about to ask his bosses for that much for such a“tricky, high-risk”venture, he says. The patent lapsed after Transgène’s parent firm decided to stop paying the fees needed to keep it active.
Meulien’s group, like the Merck team, moved to focus instead on DNA vaccines and other vector-based delivery systems. The DNA platform ultimately yielded a few licensed vaccines for veterinary applications—helping, for example, to prevent infections in fish farms. And just last month, regulators in India granted emergency approval to the world’s first DNA vaccine for human use, to help ward off COVID-19. But for reasons that are not completely understood, DNA vaccines have been slow to find success in people.
Still, the industry’s concerted push around DNA technology has had benefits for RNA vaccines, too, argues Ulmer. From manufacturing considerations and regulatory experience to sequence designs and molecular insights,“ many of the things that we learned from DNA could be directly applied to RNA”, he says.“ It provided the foundation for the success of RNA.”
Continuous struggle
In the 1990s and for most of the 2000s, nearly every vaccine company that considered working on mRNA opted to invest its resources elsewhere. The conventional wisdom held that mRNA was too prone to degradation, and its production too expensive.“It was a continuous struggle,” says Peter Liljeström, a virologist at the Karolinska Institute in Stockholm, who 30 years ago pioneered a type of‘self-amplifying’RNA vaccine.
“RNA was so hard to work with,”s ays Matt Winkler, who founded one of the first RNA-focused lab supplies companies, Ambion, in Austin, Texas, in 1989. “If you had asked me back [then] if you could inject RNA into somebody for a vaccine, I would have laughed in your face.”
The mRNA vaccine idea had a more favourable reception in oncology circles, albeit as a therapeutic agent, rather than to prevent disease. Beginning with the work of gene therapist David Curiel, several academic scientists and start-up companies explored whether mRNA could be used to combat cancer. If mRNA encoded proteins expressed by cancer cells, the thinking went, then injecting it into the body might train the immune system to attack those cells.
Curiel, now at the Washington University School of Medicine in St Louis, Missouri, had some success in mice10. But when he approached Ambion about commercialization opportunities, he says, the firm told him:“ We don’t see any economic potential in this technology.”
Another cancer immunologist had more success, which led to the founding of the first mRNA therapeutics company, in 1997. Eli Gilboa proposed taking immune cells from the blood, and coaxing them to take up synthetic mRNA that encoded tumour proteins. The cells would then be injected back into the body where they could marshal the immune system to attack lurking tumours.
Gilboa and his colleagues at Duke University Medical Center in Durham, North Carolina, demonstrated this in mice11. By the late 1990s, academic collaborators had launched human trials, and Gilboa’s commercial spin-off, Merix Bioscience (later renamed to Argos Therapeutics and now called CoImmune), soon followed with clinical studies of its own. The approach was looking promising until a few years ago, when a late-stage candidate vaccine failed in a large trial; it has now largely fallen out of fashion.
But Gilboa’s work had an important consequence. It inspired the founders of the German firms CureVac and BioNTech—two of the largest mRNA companies in existence today—to begin work on mRNA. Both Ingmar Hoerr, at CureVac, and Uğur Şahin, at BioNTech, told Nature that, after learning of what Gilboa had done, they wanted to do the same, but by administering mRNA into the body directly.
“There was a snowball effect,” says Gilboa, now at the University of Miami Miller School of Medicine in Florida.
Start-up accelerator
Hoerr was the first to achieve success. While at the University of Tübingen in Germany, he reported in 2000 that direct injections could elicit an immune response in mice12. He created CureVac (also based in Tübingen) that year. But few scientists or investors seemed interested. At one conference where Hoerr presented early mouse data, he says,“ there was a Nobel prizewinner standing up in the first row saying,‘ This is completely shit what you’re telling us here—completely shit’.”(Hoerr declined to name the Nobel laureate.)
Eventually, money trickled in. And within a few years, human testing began. The company’s chief scientific officer at the time, Steve Pascolo, was the first study subject: he injected himself13 with mRNA and still has match-head-sized white scars on his leg from where a dermatologist took punch biopsies for analysis. A more formal trial, involving tumour-specific mRNA for people with skin cancer, kicked off soon after.
Şahin and his immunologist wife, Özlem Türeci, also began studying mRNA in the late 1990s, but waited longer than Hoerr to start a company. They plugged away at the technology for many years, working at Johannes Gutenberg University Mainz in Germany, earning patents, papers and research grants, before pitching a commercial plan to billionaire investors in 2007.“ If it works, it will be ground-breaking,” Şahin said. He got €150 million in seed money.
The same year, a fledgling mRNA start-up called RNARx received a more modest sum: $97,396 in small-business grant funding from the US government. The company’s founders, biochemist Katalin Karikó and immunologist Drew Weissman, both then at the University of Pennsylvania (UPenn) in Philadelphia, had made what some now say is a key finding: that altering part of the mRNA code helps synthetic mRNA to slip past the cell’s innate immune defences.
Fundamental insights
Karikó had toiled in the lab throughout the 1990s with the goal of transforming mRNA into a drug platform, although grant agencies kept turning down her funding applications. In 1995, after repeated rejections, she was given the choice of leaving UPenn or accepting a demotion and pay cut. She opted to stay and continue her dogged pursuit, making improvements to Malone’s protocols14, and managing to induce cells to produce a large and complex protein of therapeutic relevance15.
In 1997, she began working with Weissman, who had just started a lab at UPenn. Together, they planned to develop an mRNA-based vaccine for HIV/AIDS. But Karikó’s mRNAs set off massive inflammatory reactions when they were injected into mice.
She and Weissman soon worked out why: the synthetic mRNA was arousing16 a series of immune sensors known as Toll-like receptors, which act as first responders to danger signals from pathogens. In 2005, the pair reported that rearranging the chemical bonds on one of mRNA’s nucleotides, uridine, to create an analogue called pseudouridine, seemed to stop the body identifying the mRNA as a foe17.
Drew Weissman
Few scientists at the time recognized the therapeutic value of these modified nucleotides. But the scientific world soon awoke to their potential. In September 2010, a team led by Derrick Rossi, a stem-cell biologist then at Boston Children’s Hospital in Massachusetts, described how modified mRNAs could be used to transform skin cells, first into embryonic-like stem cells and then into contracting muscle tissue18. The finding made a splash. Rossi was featured in Time magazine as one of 2010’s‘people who mattered’. He co-founded a start-up, Moderna in Cambridge.
Moderna tried to license the patents for modified mRNA that UPenn had filed in 2006 for Karikó’s and Weissman’s invention. But it was too late. After failing to come to a licensing agreement with RNARx, UPenn had opted for a quick payout. In February 2010, it granted exclusive patent rights to a small lab-reagents supplier in Madison. Now called Cellscript, the company paid $300,000 in the deal. It would go on to pull in hundreds of millions of dollars in sublicensing fees from Moderna and BioNTech, the originators of the first mRNA vaccines for COVID-19. Both products contain modified mRNA.
RNARx, meanwhile, used up another $800,000 in small-business grant funding and ceased operations in 2013, around the time that Karikó joined BioNTech (retaining an adjunct appointment at UPenn).
The pseudouridine debate
Researchers still argue over whether Karikó and Weissman’s discovery is essential for successful mRNA vaccines. Moderna has always used modified mRNA—its name is a portmanteau of those two words. But some others in the industry have not.
Researchers at the human-genetic-therapies division of the pharmaceutical firm Shire in Lexington, Massachusetts, reasoned that unmodified mRNA could yield a product that was just as effective if the right‘cap’ structures were added and all impurities were removed.“ It came down to the quality of the RNA,” says Michael Heartlein, who led Shire’s research effort and continued to advance the technology at Translate Bio in Cambridge, to which Shire later sold its mRNA portfolio. (Shire is now part of the Japanese firm Takeda.)
Although Translate has some human data to suggest its mRNA does not provoke a concerning immune response, its platform remains to be proved clinically: its COVID-19 vaccine candidate is still in early human trials. But French drug giant Sanofi has been convinced of the technology’s promise: in August 2021, it announced plans to acquire Translate for $3.2 billion. (Heartlein left last year to found another firm in Waltham, Massachusetts, called Maritime Therapeutics.)
CureVac, meanwhile, has its own immune-mitigation strategy, which involves altering the genetic sequence of the mRNA to minimize the amount of uridine in its vaccines. Twenty years of working on that approach seemed to be bearing fruit, with early trials of the company’s experimental vaccines for rabies19 and COVID-1920 both proving a success. But in June, data from a later-stage trial showed that CureVac’s coronavirus vaccine candidate was much less protective than Moderna’s or BioNTech’s.
In light of those results, some mRNA experts now consider pseudouridine an essential component of the technology—and so, they say, Karikó’s and Weissman’s discovery was one of the key enabling contributions that merits recognition and prizes.“ The real winner here is modified RNA,” says Jake Becraft, co-founder and chief executive of Strand Therapeutics, a Cambridge-based synthetic-biology company working on mRNA-based therapeutics.
Not everyone is so certain.“ There are multiple factors that may affect the safety and efficacy of an mRNA vaccine, chemical modification of mRNA is only one of them,” says Bo Ying, chief executive of Suzhou Abogen Biosciences, a Chinese company with an mRNA vaccine for COVID-19 now in late-stage clinical testing. (Known as ARCoV, the product uses unmodified mRNA.)
Fat breakthrough
As for linchpin technologies, many experts highlight another innovation that was crucial for mRNA vaccines—one that has nothing to do with the mRNA. It is the tiny fat bubbles known as lipid nanoparticles, or LNPs, that protect the mRNA and shuttle it into cells.
This technology comes from the laboratory of Pieter Cullis, a biochemist at the University of British Columbia in Vancouver, Canada, and several companies that he founded or led. Beginning in the late 1990s, they pioneered LNPs for delivering strands of nucleic acids that silence gene activity. One such treatment, patisiran, is now approved for a rare inherited disease.
After that gene-silencing therapy began to show promise in clinical trials, in 2012, two of Cullis’s companies pivoted to explore opportunities for the LNP delivery system in mRNA-based medicines. Acuitas Therapeutics in Vancouver, for example, led by chief executive Thomas Madden, forged partnerships with Weissman’s group at UPenn and with several mRNA companies to test different mRNA–LNP formulations. One of these can now be found in the COVID-19 vaccines from BioNTech and CureVac. Moderna’s LNP concoction is not much different.
The nanoparticles have a mixture of four fatty molecules: three contribute to structure and stability; the fourth, called an ionizable lipid, is key to the LNP’s success. This substance is positively charged under laboratory conditions, which offers similar advantages to the liposomes that Felgner developed and Malone tested in the late 1980s. But the ionizable lipids advanced by Cullis and his commercial partners convert to a neutral charge under physiological conditions such as those in the bloodstream, which limits the toxic effects on the body.
What’s more, the four-lipid cocktail allows the product to be stored for longer on the pharmacy shelf and to maintain its stability inside the body, says Ian MacLachlan, a former executive at several Cullis-linked ventures.“ It’s the whole kit and caboodle that leads to the pharmacology we have now,” he says.
By the mid-2000s, a new way to mix and manufacture these nanoparticles had been devised. It involved using a‘T-connector’apparatus, which combines fats (dissolved in alcohol) with nucleic acids (dissolved in an acidic buffer). When streams of the two solutions merged, the components spontaneously formed densely packed LNPs21. It proved to be a more reliable technique than other ways of making mRNA-based medicines.
Once all the pieces came together,“ it was like, holy smoke, finally we’ve got a process we can scale”, says Andrew Geall, now chief development officer at Replicate Bioscience in San Diego. Geall led the first team to combine LNPs with an RNA vaccine22, at Novartis’s US hub in Cambridge in 2012. Every mRNA company now uses some variation of this LNP delivery platform and manufacturing system—although who owns the relevant patents remains the subject of legal dispute. Moderna, for example, is locked in a battle with one Cullis-affiliated business—Arbutus Biopharma in Vancouver—over who holds the rights to the LNP technology found in Moderna’s COVID-19 jab.
An industry is born
By the late 2000s, several big pharmaceutical companies were entering the mRNA field. In 2008, for example, both Novartis and Shire established mRNA research units—the former (led by Geall) focused on vaccines, the latter (led by Heartlein) on therapeutics. BioNTech launched that year, and other start-ups soon entered the fray, bolstered by a 2012 decision by the US Defense Advanced Research Projects Agency to start funding industry researchers to study RNA vaccines and drugs. Moderna was one of the companies that built on this work and, by 2015, it had raised more than $1 billion on the promise of harnessing mRNA to induce cells in the body to make their own medicines—thereby fixing diseases caused by missing or defective proteins. When that plan faltered, Moderna, led by chief executive Stéphane Bancel, chose to prioritize a less ambitious target: making vaccines.
That initially disappointed many investors and onlookers, because a vaccine platform seemed to be less transformative and lucrative. By the beginning of 2020, Moderna had advanced nine mRNA vaccine candidates for infectious diseases into people for testing. None was a slam-dunk success. Just one had progressed to a larger-phase trial.
But when COVID-19 struck, Moderna was quick off the mark, creating a prototype vaccine within days of the virus’s genome sequence becoming available online. The company then collaborated with the US National Institute of Allergy and Infectious Diseases (NIAID) to conduct mouse studies and launch human trials, all within less than ten weeks.
BioNTech, too, took an all-hands-on-deck approach. In March 2020, it partnered with New York-based drug company Pfizer, and clinical trials then moved at a record pace, going from first-in-human testing to emergency approval in less than eight months.
Both authorized vaccines use modified mRNA formulated in LNPs. Both also contain sequences that encode a form of the SARS-CoV-2 spike protein that adopts a shape more amenable to inducing protective immunity. Many experts say that the protein tweak, tailored for coronaviruses by NIAID vaccinologist Barney Graham and structural biologists Jason McLellan at the University of Texas at Austin and Andrew Ward at Scripps, is also a prize-worthy contribution, albeit not one that is specific to mRNA vaccination, because the concept can be applied to many viral vaccines.
The lightning-fast quest for COVID vaccines—and what it means for other diseases
Some of the furore in discussions of credit for mRNA discoveries relates to who holds lucrative patents. But much of the foundational intellectual property dates back to claims made in 1989 by Felgner, Malone and their colleagues at Vical (and in 1990 by Liljeström). These had only a 17-year term from the date of issue and so are now in the public domain.
Even the Karikó–Weissman patents, licensed to Cellscript and filed in 2006, will expire in the next five years. Industry insiders say this means that it will soon become very hard to patent broad claims about delivering mRNAs in lipid nanoparticles, although companies can reasonably patent particular sequences of mRNA—a form of the spike protein, say—or proprietary lipid formulations.
Firms are trying. Moderna, the dominant player in the mRNA vaccine field, which has experimental shots in clinical testing for influenza, cytomegalovirus and a range of other infectious diseases, got two patents last year covering the broad use of mRNA to produce secreted proteins. But multiple industry insiders told Nature they think these could be challengeable.
How COVID unlocked the power of RNA vaccines
“We don’t feel there’s a lot that is patentable, and certainly not enforceable,” says Eric Marcusson, chief scientific officer of Providence Therapeutics, an mRNA vaccines company in Calgary, Canada.
Nobel debate
As for who deserves a Nobel, the names that come up most often in conversation are Karikó and Weissman. The two have already won several prizes, including one of the Breakthrough Prizes (at $3 million, the most lucrative award in science) and Spain’s prestigious Princess of Asturias Award for Technical and Scientific Research. Also recognized in the Asturias prize were Felgner, Şahin, Türeci and Rossi, along with Sarah Gilbert, the vaccinologist behind the COVID-19 vaccine developed by the University of Oxford, UK, and the drug firm AstraZeneca, which uses a viral vector instead of mRNA. (Cullis’s only recent accolade was a $5,000 founder’s award from the Controlled Release Society, a professional organization of scientists who study time-release drugs.)
Some also argue that Karikó should be acknowledged as much for her contributions to the mRNA research community at large as for her discoveries in the lab.“ She’s not only an incredible scientist, she’s just a force in the field,” says Anna Blakney, an RNA bioengineer at the University of British Columbia. Blakney gives Karikó credit for offering her a speaking slot at a major conference two years ago, when she was still in a junior postdoc position (and before Blakney co-founded VaxEquity, a vaccine company in Cambridge, UK, focusing on self-amplifying-RNA technology). Karikó“ is actively trying to lift other people up in a time when she’s been so under-recognized her whole career”.
Although some involved in mRNA’s development, including Malone, think they deserve more recognition, others are more willing to share the limelight.“ You really can’t claim credit,” says Cullis. When it comes to his lipid delivery system, for instance,“ we’re talking hundreds, probably thousands of people who have been working together to make these LNP systems so that they’re actually ready for prime time.”
“Everyone just incrementally added something—including me,” says Karikó.
Looking back, many say they’re just delighted that mRNA vaccines are making a difference to humanity, and that they might have made a valuable contribution along the road.“ It’s thrilling for me to see this,” says Felgner.“ All of the things that we were thinking would happen back then—it’s happening now.”
References
1. Malone, R. W., Felgner, P. L. & Verma, I. M. Proc. Natl Acad. Sci. USA 86, 6077–6081 (1989).
2. Malone, R. W. Focus 11, 61–66 (1989).
3. Dimitriadis, G. J. Nature 274, 923–924 (1978).
4. Ostro, M. J., Giacomoni, D., Lavelle, D., Paxton, W. & Dray, S. Nature 274, 921–923 (1978).
5. Melton, D. A. et al. Nucleic Acids Res. 12, 7035–7056 (1984).
6. Krieg, P. A. & Melton, D. A. Nucleic Acids Res. 12, 7057–7070 (1984).
7. Wolff, J. A. et al. Science 247, 1465–1468 (1990).
8. Martinon, F. et al. Eur. J. Immunol. 23, 1719–1722 (1993).
9. Jirikowski, G. F., Sanna, P. P., Maciejewski-Lenoir, D. & Bloom, F. E. Science 255, 996–998 (1992).
10. Conry, R. M. et al. Cancer Res. 55, 1397–1400 (1995).
11. Boczkowski, D., Nair, S. K., Snyder, D. & Gilboa, E. J. Exp. Med. 184, 465–472 (1996).
12. Hoerr, I., Obst, R., Rammensee, H. G. & Jung, G. Eur. J. Immunol. 30, 1–7 (2000).
13. Probst, J. et al. Gene Ther. 14, 1175–1180 (2007).
14. Karikó, K., Kuo, A., Barnathan, E. S. & Langer, D. J. Biochim. Biophys. Acta 1369, 320–334 (1998).
15. Karikó, K., Kuo, A. & Barnathan, E. Gene Ther. 6, 1092–1100 (1999).
16. Karikó, K., Ni, H., Capodici, J., Lamphier, M. & Weissman, D. J. Biol. Chem. 279, 12542–12550 (2004).
17. Karikó, K., Buckstein, M., Ni, H. & Weissman, D. Immunity 23, 165–175 (2005).
18. Warren, L. et al. Cell Stem Cell 7, 618–630 (2010).
19. Aldrich, C. et al. Vaccine 39, 1310–1318 (2021).
Susan Molineaux,는 Serial Entrepreneur입니다. Proteasome Inhibitor를 개발하는 Proteolix를 설립해서 2003년부터 2005년까지 CSO였다가 2005년부터 2009년까지 President & CEO였습니다. Susan Molineaux가 CEO로 있는 동안 Carfilzomib (Kryprolis, treatment of multiple myeloma)의 상용화를 이끌어내고 Onyx Pharmaceuticals에 Proteolix가 M&A 됩니다.
Proteolix, Inc. today announced that it has signed a definitive agreement to be acquired by Onyx Pharmaceuticals, Inc. (Nasdaq: ONXX). Proteolix is a privately-held biopharmaceutical company focused on discovering and developing novel therapies that target the proteasome for the treatment of hematological malignancies and solid tumors. Proteolix’s lead compound, carfilzomib, is a proteasome inhibitor currently in multiple clinical trials, including an advanced Phase 2b clinical trial for patients with relapsed and refractory multiple myeloma.
“Proteolix has succeeded in pioneering a new class of potent proteasome inhibitors, as demonstrated by the promising data achieved in multiple studies of our lead candidate, carfilzomib. We believe Onyx truly shares our vision for carfilzomib as an important new therapy in oncology and recognizes Proteolix’s scientific leadership in proteasome inhibition,” said John A. Scarlett, M.D., President and Chief Executive Officer of Proteolix. “Onyx’s proven track record and commercial resources in oncology are impressive. We are excited to join forces and together we are poised to advance carfilzomib through regulatory approval and achieve our ultimate objective of helping patients.”
Under the terms of the transaction, Onyx will make a $276 million cash payment upon closing of the transaction. Additional payments include $40 million payable in 2010 based on the achievement of a development milestone and up to $535 million contingent upon the achievement of anticipated approvals for carfilzomib in the U.S. and Europe. Of the potential $535 million, a payment of $170 million is based upon the achievement of accelerated U.S. Food and Drug Administration approval. The transaction is expected to close in the fourth quarter of 2009, subject to the receipt of clearance under the Hart-Scott-Rodino Act and customary closing conditions.
Proteolix is a leader in developing therapeutics that inhibit the cellular proteasome, a validated and well-characterized approach to treating certain hematologic cancers. Carfilzomib is the first in a new class of selective and irreversible proteasome inhibitors. To date, carfilzomib has demonstrated strong response rates in multiple studies and a potentially more tolerable safety profile than currently approved agents. An ongoing accelerated approval Phase 2b trial in patients with relapsed and refractory multiple myeloma is expected to complete enrollment in 2009 with data anticipated in the second half of 2010. Carfilzomib is also being evaluated in a companion Phase 2 trial in relapsed multiple myeloma. A Phase 3 trial evaluating carfilzomib in combination with lenalidomide and dexamethasone as a potential treatment option for patients with multiple myeloma is expected to begin in 2010. Carfilzomib is also being evaluated in a Phase 1b/2 study for solid tumor cancers. In addition, Proteolix has discovered additional next-generation proteasome inhibitors to which it holds worldwide development and commercialization rights, including an oral proteasome inhibitor and a selective immunoproteasome inhibitor.
“Treatment options in multiple myeloma have historically been limited, and there is a tremendous need to expand the treatment paradigm with agents offering an improved efficacy and safety profile,” said Michael Kauffman, M.D., Ph.D., Chief Medical Officer at Proteolix. “Carfilzomib is in multiple ongoing clinical studies and has revealed clear single-agent activity in a heavily pre-treated multiple myeloma patient population, as well as being well tolerated alone, or in combination with Revlimid. Upcoming data for carfilzomib could support the potential near-term introduction of a novel therapy for this debilitating disease.”
Susan Molineaux,는 2010년에 UCSF의 기술을 임상에 적용하기 위해 Calithera Biosciences라는 Oncology 회사를 설립하게 됩니다.
Calithera Biosciences, a company developing novel oncology therapeutics, today announced the completion of a Series A financing totaling $40 million. Morgenthaler Ventures led the financing with U.S. Venture Partners, Advanced Technology Ventures, Delphi Ventures and Mission Bay Capital also participating in the round. The capital will be used to support the company’s pioneering efforts to develop activators of caspases, the proteases that promote apoptotic cell death, for the treatment of cancer and other proliferative diseases.
“Promoting apoptosis in cancer cells is a validated approach to the treatment of cancer, as many oncology drugs on the market today are known to kill tumor cells by activating apoptotic pathways, albeit through indirect means,” said Susan Molineaux, Ph.D., co-founder and Chief Executive Officer of Calithera. “By targeting caspases directly, we hope to develop agents that have broad utility across many types of cancer, with greater specificity than current treatments and the potential to overcome chemoresistance.”
Calithera’s technology was developed by and licensed from the laboratory of co-founder James Wells, Ph.D., chair of the Department of Pharmaceutical Chemistry in the University of California, San Francisco School of Pharmacy. Dr. Wells’s laboratory has successfully identified several novel compounds that selectively activate procaspases and trigger apoptosis in cancer cells. Proceeds from the financing will be used to advance one or more caspase activators through preclinical development and into Phase 1 clinical trials in cancer patients. In parallel, the company will expand its technology for targeting allosteric activating sites to other enzymes with therapeutic potential in cancer.
“Most drug discovery efforts are focused on identifying drugs that inhibit enzyme function,” said Dr. Wells. “But, interestingly, many cellular enzymes remain dormant until activated. In the case of caspases, they can be activated on demand by mimicking the natural process with small molecules.”
“I am excited about the novel chemical approach that Calithera is taking,” said Chris Christoffersen, Ph.D., of Morgenthaler Ventures and Chairman of the Calithera Board. “The technology to discover small molecules that target binding sites to activate, rather than inhibit, enzymes has the potential to be a powerful and broadly applicable approach to developing innovative therapies across many targets.”
Expert Leadership Team in Place
The management team of Calithera brings to the company both deep scientific expertise and extensive experience in drug development.
Susan Molineaux, Ph.D., was most recently a founder and Chief Executive Officer of Proteolix, a company that developed proteasome inhibitors. Proteolix was in late-stage clinical trials with carfilzomib in multiple myeloma when Onyx Pharmaceuticals acquired the company in 2009 for $851 million. Prior to forming Proteolix, Dr. Molineaux held leadership positions at Rigel Pharmaceuticals and Praecis Pharmaceuticals. Dr. Molineaux began her career as a scientist in the Immunology group at Merck.
Mark Bennett, Ph.D., Senior Vice President of Research at Calithera, was Vice President of Research at Proteolix. Previously, he was Director of Cell Biology at Rigel Pharmaceuticals. Prior to that, Dr. Bennett served as an Assistant Professor in the Department of Molecular and Cell Biology at University of California, Berkeley.
Eric Sjogren, Ph.D., Senior Vice President of Drug Discovery at Calithera, was most recently the Vice President and Head of Medicinal Chemistry at Roche, Palo Alto. He held a series of positions during his 15-year tenure at Roche. Prior to that, Dr. Sjogren was at Syntex for eight years.
Calithera’s Board of Directors includes Susan Molineaux, Ph.D., Chris Christoffersen, Ph.D., Larry Lasky, Ph.D., from U.S. Venture Partners, Jean George from Advanced Technology Ventures, Deepa Pakianathan, Ph.D., from Delphi Ventures, and James Wells, Ph.D.
(By Dan Fost) With innovation as the watchword, a biotech spinoff from the UCSF School of Pharmacy announced a $40 million Series A round of investment last week – hailed by an investor as “one of the largest first rounds of financing in some time.” Calithera Biosciences, which launched this year out of the lab of Jim Wells, PhD, at Mission Bay, brings a novel approach to killing cancer cells that several major biotech investors see as having the potential to help speed recovery from the disease. Calithera provides the latest example of cutting-edge research and technology that have originated at UCSF and spun out into companies or industry partnerships, with the intent of having a direct, positive impact on patients’ lives. Since UCSF spawned the biotech industry in the 1970s with the launch of Genentech and the discovery of recombinant insulin as the first biotech drug, the University has issued 1,757 biomedical patents and has spun off more than 66 companies from its research. Those UCSF patents have led the UC system in generating license and royalty fees during these cash-tight times. The UCSF discovery that led to the current hepatitis B vaccine generates the largest royalties of the entire UC system, and the discovery of human growth hormone, which was developed and brought to market by Genentech, is among the top five. Although it’s not the largest UC campus, UCSF has consistently ranked first in the 10-campus system, as measured by total utility licenses issued, total utility patents and total license income. Over the last 10 years alone, the biomedical campus has issued 602 UCSF patents, averaging $64 million per year in licensing, litigation and royalty income. “We are certainly very prolific in winning US patents, and overall, we’ve been very successful in licensing,” said Joel Kirschbaum, PhD, director of the UCSF Office of Technology Management. While Kirschbaum said the licensing revenue helps support programs as well as the University’s educational mission, he noted, “Our fundamental mission is to transfer the fruits of publicly funded research to benefit the public.” The real win in cases like Calithera, he said, will come if the therapy the company develops achieves its goal of significantly helping cancer patients. Calithera’s announcement comes one month after the release of an economic impact report that UCSF commissioned, which showed that the University – through its vast research and medical enterprise – has a $6.2 billion annual economic impact on the Bay Area. The Calithera deal also stands in the vanguard of a new model that could generate faster translation of scientific research into patient care, while also increasing funding to the UC system. Among its venture backers is Mission Bay Capital (MBC), an independent venture fund that is managed by the California Institute for Quantitative Biosciences (QB3), based at UCSF’s Mission Bay campus. “In the long term, universities need to be more innovative about finding revenue streams to survive,” said Douglas Crawford, PhD, QB3’s associate director and a pro bono managing partner of Mission Bay Capital, noting that California will never return to its 1960s-level of investment in the University system. “We’ve got to be nimble about finding resources.” MBC benefits from advisers that include renowned investors Brook Byers, Chris Christoffersen, PhD, and John Wadsworth Jr. as it helps connect savvy investors with the world-class research taking place in University labs. In the past few years, UCSF has put a renewed emphasis on working with industry in an effort to expedite translational medicine, a trend that has accelerated under Chancellor Susan Desmond-Hellmann, MD, MPH, a former Genentech executive. Like Desmond-Hellmann, Wells is a veteran UCSF scientist who left the University for a long stint in industry. A member of the prestigious National Academy of Sciences, Wells spent 16 years at Genentech, and then started his own firm, Sunesis Pharmaceuticals, now a publicly traded company. He returned to UCSF in 2005 and now chairs the Department of Pharmaceutical Chemistry in the School of Pharmacy, with a joint appointment in the School of Medicine’s Department of Cellular and Molecular Pharmacology. His lab is focused on understanding and modulating signals in human cells, working with small molecules. For the past five to seven years, the lab has taken an unconventional approach to studying caspases – enzymes that kill cells – which cancer cells have been able to avoid. Most drugs work to inhibit enzyme function, but Wells’ lab has studied what would happen if the enzymes were activated instead. “Cells have within them the ability to commit suicide,” Wells said. “When they are heavily virally infected or have accrued mutations on the pathway to developing cancer, they have inborn mechanisms by which can they commit suicide for the benefit of the organism. But most cancers find a way to avoid this inborn mechanism. They create mutations that don’t allow it to happen.” Many cancer drugs try to stimulate that process of programmed cell death, known as apoptosis. Those drugs start at a point that, Wells said, is upstream from the cancer. They launch what he called a “bucket brigade” designed to cause cell death farther downstream. The problem, he said, is that “cancers figure out ways to lesion it, and avoid the bucket brigade. We thought it might be an interesting approach as an anticancer drug if we could activate caspases directly” at the downstream junction. Three years ago, a postdoctoral fellow in the lab, Dennis Wolan, PhD, used a high-throughput screen in Wells’ Small Molecule Discovery Center at QB3 to discover a compound that would activate the caspases in a cancer cell and kill it. “For reasons we don’t fully understand, but are beginning to understand, it will kill cancer cells much more rapidly than other cells,” Wells said. The lab published a paper in Science last fall, which attracted interest from venture capitalists. Teaming up with biotech industry veteran Susan Molineaux, PhD, who is co-founder and chief executive officer of Calithera, led to the formation of the company, which is based in South San Francisco. In last week’s announcement, Calithera announced the hiring of Molineaux and the rest of the management team and board of directors, which includes Wells. The $40 million funding round was led by Morgenthaler Ventures and included, in addition to Mission Bay Capital, US Venture Partners, Advanced Technology Ventures and Delphi Ventures.
Glutaminase Inhibitor인 Telaglenastat (CB-839)의 임상1상을 진행시키기 위해 $35 Million Series D를 했습니다.
Calithera Biosciences, a biotechnology company focused on the development of novel cancer therapeutics, today announced the successful completion of a $35 million Series D financing led by Adage Capital Partners, LP. Joining Adage as new investors in this financing are the Longwood Fund and two other institutional investor groups. Existing investors Morgenthaler Ventures, Advanced Technology Ventures and Delphi Ventures, who have funded the company through earlier rounds of financing, also participated in the transaction. In conjunction with this financing, Christoph Westphal, MD, PhD, Partner and Co-Founder of the Longwood Fund, will join Calithera’s Board of Directors as an observer.
Calithera will use the proceeds from the financing to accelerate the development of the company’s lead clinical-stage candidate CB-839 through Phase 1 clinical trials in patients with advanced solid and hematological tumors. CB-839 is a potent and selective orally bioavailable glutaminase inhibitor that blocks the growth and survival of many different types of cancer cells by interfering with their metabolism of glutamine. Calithera will also continue to develop its pipeline of novel therapeutics, including an inhibitor of MCL-1, an anti-apoptotic BCL-2 homolog over-expressed in many cancers.
“With this strong support from new and current investors, we will advance CB-839 through an early set of clinical trials aimed at identifying specific populations of cancer patients with glutamine-dependent tumors that would benefit from treatment with CB-839,” said Susan Molineaux, PhD, President and Chief Executive Officer of Calithera Biosciences. “CB-839 has demonstrated promising anti-tumor activity in a broad range of tumor types that depend on glutaminase for cell growth and survival. We look forward to initiating clinical studies for CB-839.”
“The Calithera team possesses an impressive track record in discovering and developing successful oncology drugs,” said Christoph Westphal. “Calithera’s drug, CB-839, is an important new therapeutic candidate that has the pharmacological characteristics necessary to compete as a successful commercial oncology product. We look forward to CB-839 and additional innovative pipeline candidates entering clinical trials and making significant contributions to the advancement of cancer therapy.”
Calithera Biosciences, Inc. (Nasdaq: CALA), a clinical stage biotechnology company focused on discovering and developing novel small molecule drugs for the treatment of cancer and other life-threatening diseases, today announced the closing of its previously announced public offering of 5,750,000 shares of common stock, including 750,000 shares sold pursuant to the underwriter’s exercise in full of its option to purchase additional shares. Gross proceeds from the offering at a public offering price of $6.25 per share, before underwriting discounts and commissions and offering expenses, were approximately $36 million. Citigroup acted as the sole book-running manager for the offering.
그러나 Glutaminase Inhibitor인 Telaglenastat (CB-839)의 임상2상 중 하나였던 CANTATA 임상이 primary end point를 맞추는 데 실패함에 따라 35%의 인력 구조조정을 하게 됩니다. 여기서부터 회사가 위태로워지기 시작합니다. 그래도 이 때까지는 $115 Million이나 현금이 있는 상태여서 개발에 힘을 실을 시간은 있었던 것이 아닌가 싶습니다. 하지만 이후 경영진은 Caspase Hypothesis를 포기하고 Takeda의 두신약에 매달리게 됩니다. 그리고 엎친데 덮친 격으로 코로나 팬데믹으로 인해 임상 진행도 쉽지 않았던 것 같습니다.
A phase 2 clinical trial of Calithera Biosciences’ telaglenastat in renal cell carcinoma has missed its primary endpoint. The failure of the glutaminase inhibitor to improve progression-free survival (PFS) prompted Calithera to lay off 35% of its staff to stretch its cash reserves beyond future readouts.
Telaglenastat is designed to stop cancer cells from consuming the glutamine they need to survive and grow. Calithera tested the hypothesis by randomizing 444 metastatic renal cell carcinoma patients to receive telaglenastat or placebo orally twice a day, in addition to Exelixis’ Cabometyx, and assessing the effect of the experimental drug on PFS.
The drug flunked the test. Median PFS in the telaglenastat arm was 9.2 months, compared to 9.3 months in the control cohort. Almost two-thirds of patients had previously been treated with PD(L)-1 therapies. Calithera said the arms were well balanced.
Calithera responded to the setback in the CANTATA clinical trial by outlining plans to lay off 35% of its employees. With Calithera having 90 full-time employees at the last publicly disclosed count, the proposal suggests around 30 positions are at risk. The cuts are intended to stretch the $115 million Calithera had in the bank at the end of 2020 past data drops from two clinical trials.
One of the studies, KEAPSAKE, is assessing telaglenastat in KEAP1/NRF2 mutant non-small cell lung cancer (NSCLC) patients. Calithera is continuing the NSCLC trial despite the clear failure of CANTATA in the belief that there is a strong, distinct rationale for targeting the patient population.
The KEAP1/NRF2 pathway’s regulation of reactive oxygen species is implicated in the development of some cases of NSCLC. As the process results in cells that depend on glutaminase activity, Calithera sees reasons to think telaglenastat can succeed in a genetic subset of NSCLC patients despite failing in renal cell carcinoma.
Laying off the staff is intended to enable Calithera to keep going into 2022, by which time it will have delivered interim results from the NSCLC clinical trial. The extended cash runway also takes Calithera past the completion of a phase 1 clinical trial of arginase inhibitor CB-280 in cystic fibrosis patients.
The readouts could revitalize Calithera, but, for now, the company is at a low point. Shares in the West Coast biotech fell more than 50% in premarket trading, sinking to their lowest point since its 2014 IPO.
Calithera Biosciences, Inc. (Nasdaq: CALA), a clinical-stage, precision oncology biopharmaceutical company, today announced an agreement with Takeda Pharmaceutical Company Limited (“Takeda”) to acquire two clinical-stage compounds, both of which have demonstrated single-agent clinical activity with the greatest potential in biomarker-defined cancer-patient populations. The compounds, sapanisertib (CB-228, formerly TAK-228) and mivavotinib (CB-659, formerly TAK-659), further strengthen Calithera’s pipeline of clinical-stage targeted therapies.
“We believe that these clinical-stage compounds are an excellent complement to our internally-developed pipeline programs, and fit well with our current strategic focus on biomarker-driven therapeutic approaches. We are encouraged by the promising single-agent clinical data that suggest these investigational therapies could help transform treatment for multiple cancer patient populations with high unmet need,” said Susan Molineaux, PhD, president and chief executive officer of Calithera. “Specifically, sapanisertib has the potential to be the first targeted treatment for patients with NRF2-mutated squamous non-small cell lung cancer. We have learned a great deal about the unmet medical need of patients with KEAP1/NRF2 mutations, as well as how to identify and recruit these patients, during the conduct of our KEAPSAKE trial evaluating telaglenastat. This complementary approach in KEAP1/NRF2-mutant squamous NSCLC demonstrates our commitment to these patients and the pathway.
“Additionally, mivavotinib has the potential to be a best-in-class SYK inhibitor in non-Hodgkin’s lymphoma, as well as a first-to-market approach for patients with diffuse large B-cell lymphoma whose tumors harbor MyD88 and/or CD79 mutations.
“We plan to start a clinical trial in squamous NSCLC with sapanisertib and a clinical trial in DLBCL with mivavotinib, both in biomarker specific populations, and generate data in the next 12 to 18 months that will define the clinical development and potential regulatory approval paths for both of these compounds.”
The terms of the transaction include a total upfront cash payment to Takeda of $10 million and $35 million issued to Takeda in Calithera Series A preferred stock. Additionally, Takeda will be eligible to receive from Calithera clinical development, regulatory and sales milestone payments across both programs. Calithera will pay tiered royalties of high single-digits to low teens on future net sales should these candidates achieve regulatory approvals and subsequent commercial availability.
“Collaboration is an important aspect of our R&D strategy and at the center of our efforts to deliver new treatment options to patients. We are confident that Calithera, with their highly capable and experienced team, is the ideal partner to resume the development of sapanisertib and mivavotinib, and to maximize their potential to address underserved patient populations,” said Christopher Arendt, Ph.D., head of Oncology Cell Therapy and Therapeutic Area Unit of Takeda. “We look forward to seeing how these programs advance under Calithera’s leadership.”
Sapanisertib is a dual TORC 1/2 inhibitor that targets a key survival mechanism in KEAP1/NRF2-mutated tumor cells. These mutations are found in a considerable sub-population of patients across multiple solid tumor types. Sapanisertib has demonstrated promising single-agent activity in patients with relapsed/refractory NRF2-mutated squamous non-small cell lung cancer (NSCLC) and exhibits differential anti-tumor activity compared to rapalog inhibitors of TORC1 in NRF2-mutant squamous NSCLC in vivo models. A Phase 2 study planned to begin in the first quarter of 2022 will further evaluate sapanisertib as a monotherapy in patients with squamous NSCLC harboring a NRF2 mutation.
Mivavotinib is a SYK inhibitor that targets the constitutively active BCR pathway in many non-Hodgkin’s lymphoma (NHL) cases as well as the constitutively active inflammatory signaling pathway in MyD88-mutated NHL. In early phase studies, mivavotinib showed promising single-agent responses in relapsed/refractory diffuse large B-cell lymphoma (DLBCL). In addition, recent preclinical studies have shown enhanced SYK activity and sensitivity to SYK inhibition in DLBCL and other NHLs harboring mutations in MyD88 and/or CD79, which comprise a distinct genetic subset of DLBCL known to have poor outcomes with standard-of-care therapy. Accordingly, Calithera plans to initiate a Phase 2 study of mivavotinib in 2022 for the treatment of patients with DLBCL with and without mutations in MyD88 and CD79. Beyond DLBCL, both preclinical and clinical data support expansion across additional NHL subtypes and other hematologic malignancies as part of long-term plans.
Calithera Biosciences has reported that it will suspend the Phase II KEAPSAKE clinical trial of telaglenastat in stage IV non-squamous non-small cell lung cancer (NSCLC) due to a lack of clinical benefit.
This placebo-controlled, randomised, double-blind trial analysed the safety and anti-tumour activity of telaglenastat in combination with standard-of-care chemoimmunotherapy as front-line treatment for stage IV NSCLC patients. These subjects had tumours with Kelch Like ECH Associated Protein 1 (KEAP1) or Nuclear factor-erythroid factor 2-related factor 2 (NRF2) mutations. The trial had randomised a total of 40 subjects when it was unblinded on 27 October 2021. The efficacy results, which included investigator-evaluated progression-free survival, did not show any clinical benefit. Interim analysis findings of the trial also showed a reduced likelihood of an eventual positive outcome.
Calithera Biosciences has kept its head down since a phase 2 fail in 2021 caused layoffs for a third of its workforce. But in the wake of enrollment delays for two lead cancer therapies, the cash-strapped biotech has given up the fight and announced it’s liquidating the business.
“The board of directors and management devoted substantial time and effort in identifying and pursuing various opportunities, but we were unable to complete a transaction that would allow us to continue the development of our clinical programs and enhance shareholder value,” CEO Susan Molineaux, Ph.D., said in a release Monday.
As a result, all clinical programs will be shut down, with “most” of the company’s employees terminated by the end of this quarter.
“Importantly, I would like to sincerely thank our employees and others who have supported Calithera over the years,” Molineaux added in her statement yesterday. “We appreciate your partnership and participation, and we truly wish the outcome was different today.”
Money had been tight at the biotech for a while. Calithera had cash and equivalents of $34.1 million at the end of September, which was only expected to last into the second quarter of this year. In November, the company revealed it was “evaluating all options for its programs, including strategic collaboration or licensing agreements and actively considering the sale of certain programs, in order to extend its cash runway.”
It can’t have helped that things weren’t quite going to plan for its lead programs, either. Calithera also used its November financial report to reveal that “site activation delays” meant that enrollment for trials of sapanisertib and mivavotinib had been “slower than anticipated.” Still, the company said at the time that initial data from these studies were expected in mid-2023, with sapanisertib having secured an FDA fast-track tag for non-small cell lung cancer in October.
The biotech licensed both drugs from Takeda in 2021 after the failure of its own drug telaglenastat in a midstage kidney cancer test prompted Calithera to wield the ax on 35% of staff.
Calithera’s announcement is just the latest in a string of recent closures for struggling biotechs, with Otonomy winding down in December, followed by Nabriva Therapeutics last week.
2022년 3월의 파이프라인은 2021년과 완전히 달라져 버렸습니다. 저는 2021년부터 2022년 사이의 결정이 잘못된 것이 아닌가하고 생각합니다. 이해하기 힘든 것은 왜 CD73 Inhibitor인 CB-708의 임상을 시도하지 않았는가 하는 것입니다. 전임상 데이타로 봤을 때는 Best-in-class 약물이었습니다. 2019년 STIC에서 발표한 포스터입니다.
CD73 (ecto-5′-nucleotidase) has emerged as an attractive target for cancer immunotherapy of many cancers. CD73 catalyzes the hydrolysis of adenosine monophosphate (AMP) into highly immunosuppressive adenosine that plays a critical role in tumor progression. Herein, we report our efforts in developing orally bioavailable and highly potent small-molecule CD73 inhibitors from the reported hit molecule 2 to lead molecule 20 and then finally to compound 49. Compound 49 was able to reverse AMP-mediated suppression of CD8+ T cells and completely inhibited CD73 activity in serum samples from various cancer patients. In preclinical in vivo studies, orally administered 49 showed a robust dose-dependent pharmacokinetic/pharmacodynamic (PK/PD) relationship that correlated with efficacy. Compound 49 also demonstrated the expected immune-mediated antitumor mechanism of action and was efficacious upon oral administration not only as a single agent but also in combination with either chemotherapeutics or checkpoint inhibitor in the mouse tumor model.
2023년 5월에 Susan Molineaux는 Para Therapeutics를 새로 창업했습니다. Proteolix의 성공과 Calithera의 실패로 부터 배운 Susan Molineaux의 새로운 도전에 또 새로운 기대를 해 봅니다.
Off-the-shelf Cell Therapy는 꿈의 치료제라고 할 수 있는데 특히 Neurology 분야에서는 더욱 어렵지만 만일 할 수 있다면 환자들에게 정말 좋은 뉴스라 할 수 있습니다. 예를 들면 파킨슨씨병의 경우 Neuron의 Dopamine 분비에 이상이 생긴 것인데 이를 위한 회사 중 Autologous Stem Cell Therapy for Parkinson’s Disease인 “BlueRock Therapeutics”에 대해 블로그를 적은 적이 있습니다.
반대로 Kenai Therapeutics는 Allogeneic Stem Cell Therapy for Parkinson’s Disease”회사로서 iPSC-induced Stem Cell로 Doparminergic Neuron을 주입하는 치료법을 개발하는 것이 목표입니다. 이 회사의 CMO인 Howard Federoff 교수는 University of California at Irvine의 Vice Chancellor였기도 하고 Nasdaq 상장사인 Brooklyn ImmunoTherapeutics의 President & CEO였습니다. 하지만 그는 2022년 5월 새로운 벤처를 위해 사임합니다. 그 벤처가 바로 Kenai Therapeutics 인 것이죠.
2020년에 Howard Federoff교수는 하버드 대학교 김광수 교수의 Autologous Stem Cell Therapy for Parkinson’s Disease에 대한 New England Journal of Medicne의 논문과 관련한 글을 통해서 Autologous vs Allogeneic에 대해 평한 것이 있는데요. Redosability와 면역억제제를 복용할 필요가 없다는 점에서 Autologous Stem Cell Therapy가 보다 유용할 것으로 글을 썼습니다.
Cell therapies hold great promise to treat Parkinson’s disease, but there’s an ongoing debate over how to best do this. One group—and I am firmly in this camp—maintains an autologous therapy, derived from a patient’s own cells, is the correct path. Others believe an allogeneic approach, using donor cells, would be more cost-effective.
A recent study, published in the New England Journal of Medicine, added fuel to the fire. A research team, led by Harvard’s Kwang-Soo Kim, PhD, harvested cells from a person suffering from Parkinson’s disease, created induced pluripotent stem cells (iPSCs) and differentiated them into dopamine-producing neurons, which were ultimately transplanted into that person’s brain.
While this work generated some controversy, the procedure was well-tolerated for the study’s only participant, and that’s a first step towards establishing a safe track record for autologous neuronal cell transplants.
This narrow proof-of-concept will do little to settle the ongoing debate between allogeneic and autologous transplants. However, as the discussion moves forward, we must carefully assess all the evidence. This choice is an important inflection point for people with Parkinson’s disease—we need to make the right decisions based on the best, currently available data.
Autologous vs. allogeneic
Given the evidence, autologous therapies offer the most compelling opportunities to treat Parkinson’s disease, and give patients better quality of life, because they do not precipitate an immune response. Since allogeneic cells come from a donor and not from the person being treated, they are like heart, lung or kidney transplants: Recipients must take immunosuppressive drugs for their grafts to survive.
The jury is still out on which immunosuppressive regimens will be needed to protect allogeneic transplants from the immune system and how long those will be needed. Immunosuppression has a dramatic impact on quality of life, making people more vulnerable to opportunistic infections. The COVID-19 pandemic is an ongoing reminder that being immunosuppressed, for any amount of time, can be quite dangerous.
Since autologous transplants are generated from a person’s own cells, they will not need any immunosuppression, and that would be a big win for patients.
There’s also evidence that autologous neurons create synapses more efficiently. Presynaptic axons must scan their environments to make the appropriate connections with dendrites from other neurons. They perform this feat with such accuracy that many researchers believe axons and dendrites are encoded in some way to make those correct connections.
Alternatively, dopaminergic and other neurons can form autapses, in which they connect to themselves rather than targeting the right neurons. Though we do not fully understand how this mechanism works, we do know that neurons from autologous transplants share the same coding with their neighbors. As a result, they appear to reject autapses, favoring synapses and generating optimal connections to produce functional circuits. This may seem like a subtle detail, but it’s critically important if we want to restore function in Parkinson’s disease patients.
Another potential issue is redosing. We don’t know how long any cell transplant–autologous or allogeneic—will benefit certain patients. Parkinson patients with the sporadic (non-familial) form of the disease can start developing symptoms before they turn 50. As a result, they may need a second, or possibly even a third, transplant procedure to manage their disease.
This is an important consideration when choosing which path is the best choice for patients. If they have received an allogeneic transplant, that original cell line will have become immunogenic. However, patients who benefit from autologous transplants can continue to receive their own cells without any concerns over immunogenicity. For patients who need additional dopamine-producing neurons down the road, autologous cells are clearly the better option.
Making the fine distinctions
When assessing autologous and allogeneic transplants, we must recognize that there is significant diversity in these methods, and the scientific community should not treat them as two monolithic approaches.
For example, researchers can adopt various methods to convert mature cells into iPSCs and differentiate them into dopaminergic neurons for transplant. Kwang-Soo Kim’s group used a flavanol to destroy senescent cells during the differentiation process. This added another step that exposes transplant cells to a chemical, and that may not be the best way to proceed.
There are also different types of Parkinson’s disease, and treatments should reflect that diversity. Patients with sporadic Parkinson can receive transplanted dopaminergic neurons without any additional molecular interventions.
Patients with familial Parkinson’s disease, such as those who have mutations in their GBA gene, may be better served by autologous neuronal transplants that also deliver gene therapy. Providing patients with healthy genes, which produce appropriate amounts of the GBA enzyme, could go a long way towards restoring function.
These are just a couple examples of how complex it can be to develop cell therapies for Parkinson’s disease. This complexity should always be taken into account when assessing new therapies; apples should always be compared to apples.
Calculating cost and value
Sophisticated therapies and cost concerns often go together. On the surface, allogeneic neuron transplants would project as being less expensive than autologous therapies. Because a given cell line could serve a relatively large population of affected people, manufacturing costs could be reduced. However, there are other variables to consider.
While it’s true allogeneic lines could be scaled up effectively, there are risks associated with higher volumes. Larger scale can translate into greater risks of generating mutations in these cell lines. Some of these mutations could activate oncogenes.
Manufacturers would have to invest in elaborate quality control measures to protect patients, and that would add significant costs to the overall process.
Immunosuppression would also be costly. These drugs are expensive, and no one currently can say how long people will need them. Being vulnerable to infection generates other issues. For high-risk transplant patients, routine viral infections could lead to precautionary hospitalizations, as well as outpatient visits. For others, health-related complications from immunosuppression could lead to lengthy, expensive hospitalizations. The longer people need immunosuppression, the greater their risk.
We must also remember that cost projections to produce autologous neurons are moving targets, and they have a tendency to move lower. As we perfect iPSC protocols, and transfer those approaches from the lab to manufacturing, we will continue to automate processes and identify other efficiencies. By implementing the most innovative manufacturing protocols, companies can significantly reduce the cost of producing autologous neurons.
If we factor in all these expenses, the cost advantages associated with allogeneic transplants tend to dissipate. Still, costs are not exclusively a monetary realm. Some have expressed concerns that producing neurons for autologous transplant will take too long, and patients may decline precipitously while waiting.
This is actually a red herring. Mature cells can be converted into iPSCs and subsequently differentiated into dopaminergic neurons in less than four months. During that short period, it’s unlikely that a Parkinson patient eligible for this type of therapy would experience any clinically-detectable deterioration.
We still have a long way to go with cell therapies. To move forward with autologous neuron transplants, we need to approach them in a rigorous and systematic way, conduct robust, well-designed clinical trials to study safety, tolerability and efficacy and develop innovative neurosurgical approaches.
그러나, 그의 연구는 Augologous가 아닌 Allogeneic iPSC-derived dopamine progenitor cell Therapy였는데 2022년 NPJ Regenerative Medicine에 연구결과를 발표합니다.
This news came shortly after Memorial Day. Howard Federoff, chief executive of Brooklyn ImmunoTherapeutics, leaves. As announced by Brooklyn ImmunoTherapeutics Inc. in a news release and in a regulatory filing published on Tuesday, May 31, 2022, Howard J. Federoff has left his post as chief executive officer at the biopharmaceutical company, after about a year in the role, effective May 26, 2022.
Howard Federoff’s duties as CEO will be taken over temporarily by Matthew (Matt) Angel, most recently Chief Executive Officer at Factor Bioscience Inc., as Interim Chief Executive Officer.
The management change is explained as follows. Brooklyn ImmunoTherapeutics said: “Brooklyn ImmunoTherapeutics, Inc. (Nasdaq:BTX) (“Brooklyn” or the “Company”), a biopharmaceutical company focused on exploring the role that cytokine, gene editing, and cell therapy can have on the immune system for treating patients with cancer, blood disorders, and monogenic diseases, today announced the appointment of Matt Angel, Ph.D., Co-Founder, Chairman, and CEO of Factor Bioscience Inc., as Interim Chief Executive Officer and President. He will replace Howard J. Federoff, M.D., Ph.D., Chief Executive Officer and President, who departs to focus on building a new venture.”
Precise information regarding Howard Federoff’s future plans was not immediately available.
Kenai Therapeutics (previously Ryne Biotechnology)는 CIRM으로 부터 $4 Million grant를 받아서 RNDP-001이라는 iPSC-derived dopamine neuron progenitor 약물의 전임상을 할 수 있는 초기 자금을 확보합니다.
Ryne Biotechnology, Inc. (Ryne Bio), a therapeutics company leveraging induced pluripotent stem cell (iPSC) technology to discover and develop a platform of off-the-shelf neuron replacement therapies for neurological disorders, today announced that the California Institute for Regenerative Medicine (CIRM) has awarded the company a $4 million Clinical Stage Research Program (CLIN1) grant. This funding will enable the company to advance its lead candidate RNDP-001, an iPSC-derived dopamine neuron progenitor for the treatment of both inherited and idiopathic forms of Parkinson’s disease, through submission of an Investigational New Drug (IND) application within the next 12 months.
RNDP-001 has completed preclinical efficacy and safety studies.This CIRM award will allow Ryne Bio to finalize its IND-package including the production of GMP-grade materials to enable the evaluation of RNDP-001 in Phase 1 clinical trials for both inherited and idiopathic forms of Parkinson’s disease.
“We appreciate CIRM’s partnership in our vision to reverse degenerative conditions of the brain by developing off-the-shelf cell replacement therapies,” said Nick Manusos, Chief Executive Officer of Ryne Bio. “A dramatic shift in the standard of care for patients with neurodegenerative disease is long overdue. We are thrilled to be developing groundbreaking therapies for patients in need of better treatment options.”
Beyond RNDP-001, Ryne Bio is developing a platform of drug candidates, including next-generation, gene-modified programs that have the potential to modify and reverse disease progression in Parkinson’s disease and other moderate to severe central nervous system disorders.
“The underlying cause of Parkinson’s disease is progressive degeneration of a patient’s dopamine neurons. Ryne Bio is able to directly replace dopamine neurons that have been lost by utilizing precision manufacturing techniques,” said Howard Federoff, M.D., Ph.D., Ryne Bio’s Chief Medical Officer, scientific co-founder and Principal Investigator on the CLIN1 award.
In addition to funding from CIRM, Ryne Bio was launched and seeded in 2022 by Saisei Ventures, an emerging venture capital firm focused on building revolutionary advanced medicine companies. “The potential of off-the-shelf cell replacement therapies is on the cusp of being realized for complex and intractable disease,” said Jonathan Yeh, Managing Partner of Saisei Ventures. “This funding decision from CIRM provides robust validation of the Ryne Bio approach, and supports the delivery of this best-in-class therapy to patients.”
그리고 1년 후 $82 Million Series A를 통해서 RNDP-001의 임상 준비에 박차를 가할 수 있는 자금을 확보하게 됩니다. 보스턴에 있는 Fujifilm Cellular Dynamics에서 연구와 GMP 생산을 하기로 계약이 되어 있고 RNDP-001을 IND filing하고 임상에 진입함과 동시에 다른 Neurological Diseases에 대해서도 파이프라인을 늘려 간다는 계획입니다. BlueRock의 Autologous Stem Cell Therapy와 함께 Kenai의 Allogeneic Stem Cell Therapy가 Parkinson’s Disease 환자들에게 희망이 되어줄 수 있을지 기대가 됩니다.
San Diego-based Kenai Therapeutics announced its arrival on the biotech scene Thursday with $82 million in Series A financing to fund an investigational asset designed to treat Parkinson’s disease.
According to Kenai, which was formerly known as Ryne Bio, the funding was co-led by Alaska Permanent Fund Corporation, The Column Group and Cure Ventures. The round also saw participation from Saisei Venture and Euclidean Capital.
The Series A is meant to help Kenai submit an IND for RNDP-001, an iPSC-derived allogenic dopamine progenitor cell therapy intended to treat idiopathic and inherited forms of Parkinson’s. Funding will also go toward the completion of Phase I trials which will start sometime this year.
Kenai said the asset has shown “robust survival, innervation, and behavioral rescue” in preclinical models.
“We are grateful for the support of a syndicate of leading life science investors and a team of industry veterans, including scientific co-founders Dr. Howard Federoff and Dr. Jeffrey Kordower, who see the promise in Kenai’s approach to treating central nervous system disorders,” Kenai CEO Nick Manusos said in a statement. “Their guidance will be invaluable as we soon advance our lead candidate, RNDP-001, into the clinic to treat Parkinson’s disease.”
In addition to the Parkinson’s candidate, Kenai is pursuing a pipeline of off-the-shelf dopamine neuron replacement cell therapy assets targeting neurological disorders. However, no other specific disease targets were revealed in the announcement. Kenai is using Fujifilm Cellular Dynamics for its manufacturing and development services.
“Kenai’s proprietary platform leverages an emerging approach to treating central nervous system disorders by replacing neurons lost due to neurodegeneration,” Jeff Jonas, chair and board member of Kenai Therapeutics and partner at Cure Ventures, said in a statement. “The potentially curative nature of RNDP-001 for Parkinson’s disease could dramatically alter outcomes for patients with very few treatment options.”
In February 2023, when the company was known as Ryne Bio, it received a $4 million grant from the California Institute for Regenerative Medicine to support the development of RNDP-001.
RNA-level의 Gene Regulation을 Small molecule로 하는 회사들 중에서 Expansion Therapeutics에 대해 글을 쓴 적이 있습니다. Expansion 의 경우는 RNA-Small Molecule Direct Binding에 의해 단백질 합성을 조절하는 메카니즘입니다.
Accent Therapeutics는 RNA-Modifying Proteins (RMPs)를 표적함으로써 RNA level Gene Regulation을 하려는 회사로서 2018년에 Epizyme founder이자 CEO 였던 Robert Copeland 박사가 CSO이면서 Stanford University의 Howard Chang 교수와 University of Chicago의 Chuan He 교수와 함께 공동으로 창업을 한 후 시리즈 A $40 Million을 받았습니다.
Accent Therapeutics, a biopharmaceutical company developing breakthrough treatments for cancer patients, today announced $40 million in Series A capital to establish a discovery platform and pipeline of therapeutic candidates targeting RNA-modifying proteins (RMPs), a novel target space for precision cancer therapies. The Column Group, Atlas Venture and EcoR1 Capital provided the investment.
Accent Therapeutics was established to create innovative therapeutics in the rapidly advancing area of epitranscriptomics – the role of RNA structure, stability, function and translation in cell biology. Recent studies have linked certain human cancers to the activity of particular RMPs, providing a rich new target space for drug development. The Accent Therapeutics team includes seasoned drug developers, with an established record of translating novel science into innovative therapies. Leaders of that team have recently published a peer-reviewed overview of advances in the field in Nature Reviews Drug Discovery entitled “RNA-Modifying Proteins as Anticancer Drug Targets” (doi:10.1038/nrd.2018.71).
Accent’s founders include Howard Y. Chang, M.D., Ph.D. of Stanford University, Chuan He, Ph.D. of the University of Chicago and Robert A. Copeland, Ph.D., President and Chief Scientific Officer of Accent Therapeutics, who together bring broad and deep expertise in the emerging biology of epitranscriptomics, its role in human diseases and the translation of novel science to cancer drug discovery and development. “Epitranscriptomics opens a rich new target space, including RMPs that are associated with specific cancers, many with poor patient prognoses,” said Dr. Copeland. “We plan to treat patients by precisely targeting cancers that are uniquely dependent on these specific RMPs.”
“There is great value in targeting the molecular mechanisms that can go awry and drive specific cancers. We are excited to support Accent as they develop efficacious and truly differentiated cancer therapeutics,” said Larry Lasky, PhD., Partner at The Column Group and a member of the Accent Therapeutics board of directors.
“The Accent team is anchored by experienced drug developers with a track record of success in the creation of innovative precision therapies. They are well-suited to undertake the development of effective new therapies targeting RMPs,” said Jason Rhodes, Partner at Atlas Venture and a member of the Accent Therapeutics board of directors.
2년 후에 METTL3와 ADAR1 Programs을 발굴하면서 $63 Million Series B를 했는데 Abbvie Ventures와 Google Ventures 등이 새로 참여를 했습니다.
Accent Therapeutics, a biopharmaceutical company developing breakthrough treatments for cancer patients, announced today that it has completed a $63 million Series B financing. The Series B was led by EcoR1 Capital with participation by GV, AbbVie Ventures, The Mark Foundation for Cancer Research, NS Investment and Droia Ventures as well as existing investors, Atlas Venture and The Column Group.
Proceeds from the financing will be used to advance the development of Accent’s novel therapies targeting RNA-modifying proteins (RMPs), including its lead programs METTL3 and ADAR1, and to continue to expand its pipeline in the rich target space of RNA modification.
“We are thrilled to have the support of this remarkable group of investors that share our vision for developing novel therapies for patients in need,” said Shakti Narayan, Chief Executive Officer of Accent Therapeutics. “With the progress we have made to-date and expect to make in the coming months, the next phase of Accent’s growth is set to be truly transformational.”
Since launching in 2018, Accent has advanced a broad pipeline of programs, including its two lead programs – METTL3 and ADAR1. METTL3 is an RNA methyltransferase implicated in AML, specific solid tumors and immuno-oncology. ADAR1 is an RNA editor with compelling validation for solid tumors with elevated intrinsic Type I interferon-stimulated gene signaling (comprising ~15-30% of solid tumors) and has also been suggested to play a key role in immuno-oncology. By targeting the proteins that modify RNA, Accent is able to apply the proven approach of enzyme-directed small molecule therapies to a rich and novel class of enzymes with the ability to impact RNA pathobiology.
“Opportunities to have such a broad impact in novel areas of biology are becoming increasingly rare,” said Oleg Nodelman, Founder and Managing Director of EcoR1 Capital. “The team at Accent is well-positioned to lead this area of drug development and achieve the rich therapeutic potential of these exciting programs.”
시리즈B를 한 지 몇달 안되어 AstraZeneca와 $55 Million Upfront를 포함 총 $1.1 Billion 규모의 공동연구계약을 했습니다. Accent는 전임상부터 임상1상까지를 맡고 AstraZeneca는 그 이후부터 상용화까지를 맡는 계약이었습니다.
AstraZeneca will partner with Accent Therapeutics to discover, develop, and commercialize cancer therapeutics based on Accent’s RNA-modifying protein (RMP) inhibition technology, the companies said today, through a collaboration that could generate more than $1.1 billion for the Lexington, MA, biopharma.
The partnership is intended to combine AstraZeneca’s expertise in oncology drug development with Accent’s focus on small-molecule, RMP-targeting precision cancer therapeutics. Since its launch in 2018, Accent has developed two lead candidates: METTL3 is an RNA methyltransferase implicated in AML, specific solid tumors, and immuno-oncology; and ADAR1, an RNA editor with compelling validation for solid tumors with elevated intrinsic Type I interferon-stimulated gene signaling.
By targeting proteins that modify RNA, Accent reasons, it can effectively apply the proven approach of enzyme-directed small molecule therapies to a new class of enzymes with the ability to impact RNA pathobiology.
Under its collaboration with AstraZeneca, Accent has agreed to oversee R&D activity for a nominated preclinical program through Phase I clinical trials. Upon completion of Phase I, AstraZeneca has agreed to lead development and commercialization activities for the nominated program, with Accent having the option to jointly develop and commercialize with AstraZeneca in the U.S.
AstraZeneca will also have the exclusive option to license worldwide rights to two further preclinical discovery programs, for which Accent will conduct certain preclinical activities.
“A compelling area”
“The promise of RMP inhibition is a compelling area of exploration for AstraZeneca. With this collaboration, we will seek to identify novel targets and unlock the full potential of our medicines,” José Baselga, executive vice president, Oncology R&D, AstraZeneca, said in a statement.
AstraZeneca has agreed to pay Accent $55 million upfront. If Accent elects to jointly develop the nominated program, AstraZeneca would pay Accent an additional up to $1.1 billion in option fees and payments tied to achieving milestones across all programs, as well as tiered royalties on net sales ranging from mid-single digit to low-double digits.
Both companies agreed to split profits and losses in the U.S.
“This collaborative effort will enable us to rapidly advance and achieve the rich therapeutic potential of these exciting programs,” added Accent CEO Shakti Narayan, PhD, JD. “This collaboration leverages both AstraZeneca’s vast cancer expertise and resources and Accent’s rich pipeline of RMP therapeutic programs to bring new and potentially life-changing medicines to patients.
Accent’s collaboration with AstraZeneca comes less than two months after Accent completed a $63 million Series B financing.
The Series B was led by EcoR1 Capital with participation by GV, AbbVie Ventures, The Mark Foundation for Cancer Research, NS Investment and Droia Ventures as well as existing investors, Atlas Venture and The Column Group.
At the time, Accent said proceeds from the financing would be used to advance its development of RMP-inhibiting therapeutics, including METTL3 and ADAR1, and to continue expanding expand its RNA modification pipeline.
1년 후에는 Ipsen에서 METTL3 프로그램을 총 $446 Million 및 Sales royalties를 포함하는 계약으로 인수했습니다.
Ipsen (Euronext: IPN; ADR: IPSEY) and Accent Therapeutics (Accent) have signed an exclusive worldwide-collaboration agreement to research, develop, manufacture, and commercialize Accent’s pre-clinical stage METTL3 program.
Acute myeloid leukemia (AML) is a difficult to treat cancer of the blood and bone marrow, accounting for a third of all new cases of leukemia in the US each year.1 Globally, the incidence of AML has been increasing year on year across the last 20 years.2 RNA modifying proteins (RMPs) are an emerging target class that control multiple aspects of RNA biology and represent a new approach for the potential treatment of various cancers. METTL3 is an RMP that has been validated pre-clinically as a novel therapeutic target for AML.1,3 This collaboration combines Accent’s expertise in RMP-targeting therapeutics with Ipsen’s capabilities and proven track record in Oncology medicine development and commercialization.
Christelle Huguet, Senior Vice President, Head of Research, External Innovation and Early Development, Ipsen, said “Oncology is a key focus area for Ipsen as we grow our pipeline. We are delighted to partner with Accent to progress the METTL3 program as we continue our expansion into hematologic oncology. Our teams are steadfast in our commitment to areas of high unmet medical need including rare cancers, so this collaboration is strongly aligned with Ipsen’s mission and strategy for growth.”
Shakti Narayan, Chief Executive Officer of Accent Therapeutics said “This collaboration blends Ipsen’s commitment to developing and commercializing transformative oncology medicines with Accent’s leading expertise in the field of RNA modification. As we focus on developing our rich pipeline of novel RMP-targeted therapies, we are pleased to entrust our METTL3 program to the innovative team at Ipsen to bring this novel investigational therapy to patients in need.”
Under the agreement, Ipsen will pay up to $446m, comprising upfront payment as well as pre-clinical, clinical, regulatory, and sales-based milestone payments, plus tiered sales royalties ranging from mid-single digits to low-double digits.
2023년말에 Precision Medicine Online을 통해 그동안의 Accent Therapeutics의 프로그램에 대해 전체적으로 정리하는 기사가 있었습니다.
설립 초기부터 RNA modification을 하는 수백개의 유전자를 발굴하고 이 중에서 항암효과를 가진 유전자를 Knock-out 방법으로 발굴해서 1,500개의 RNA-binding protein을 찾았고 이 중에서 Precision Oncology Targets로 가장 좋은 10여개의 단백질 표적을 발굴했습니다.
Lead Program은 DHX9인데 Colorectal cancer를 포함해서 암치료제로서의 가능성에 대해 2024년 후반에 IND Filing을 하고 2025년에 임상을 시작한다는 계획입니다.
전임상 단계인 ADAR1 프로그램은 Checkpoint Inhibitors에 듣지 않는 Head and Neck Cancer, NSCLC 환자를 대상으로 하고 초기연구 중인 RNA exonuclease XRN1과 또다른 전임상 프로그램으로 Ovarian, Triple-Negative Breast, Small Cell Lung, Colorectal Cancer 환자들을 위한 표적 프로그램이 있습니다. 또한 Ipsen에 Lience-Out한 METTL3은 Acute Myeloid Leukemia 프로그램입니다.
Accent Therapeutics is advancing the first of its pipeline drugs targeting RNA-modifying proteins into investigational new drug (IND)-enabling studies in a range of cancers including tumors with microsatellite instability (MSI-high).
Accent’s lead small molecule therapeutic program inhibits DHX9, an RNA helicase that binds and unwinds double-stranded RNA and DNA. It stabilizes the genome through regulation of cellular processes such as DNA replication, transcription, translation, and RNA processing and transport. Loss of DHX9 interferes with DNA replication and increases genome instability.
Accent Founder and CSO Robert Copeland explained that DHX9’s role in stabilizing the genome is what could make DHX9 inhibitors effective against MSI-high cancers, which comprise a subset of colorectal, uterine, gastric, and other malignancies. The Lexington, Massachusetts-based company is expecting to submit an investigational new drug application for the anti-DHX9 drug in the second half of 2024 and begin clinical trials in late 2024 or early 2025.
“To the best of our knowledge, we’re the only company that has small molecule inhibitors of [DHX9] and is pursuing that as a precision cancer therapeutic,” Copeland said.
Accent launched in 2018 with $40 million in Series A financing and secured another $63 million in a Series B round in April 2020. The company’s mission is to discover and develop therapeutics targeting modifications in post-transcriptional RNA — a new field analogous to epigenetics known as epitranscriptomics. These modifications affect gene expression through regulation of RNA stability, localization, and functional efficiency. In cancer, disruption of the epitranscriptome has been implicated as a driver of tumor growth and drug resistance.
Copeland founded Accent with Howard Chang, a professor of cancer genomics at Stanford University, and Chuan He, a professor of biochemistry at the University of Chicago. Before Accent, Copeland was a founder and CSO at Epizyme, where Copeland guided the development of Tazverik (tazemetostat), an inhibitor of the histone-modifying protein EZH2 approved in the US for non-Hodgkin lymphoma and epithelial sarcoma, and numerous other clinical programs involving proteins that modify DNA.
“In 2017, I was approached by the Column Group about starting a new company that would be focused not on chromatin and histone modifications but instead RNA modifications,” Copeland said. However, Copeland added, “it’s very difficult to have potent selective small molecules that will bind directly to RNA.”
Instead, Accent’s approach is to target the proteins involved in modifying RNA. “It’s a novel area of medicine in targeting RNA pathobiology, [but] we’re doing it through a very well-established mechanism of small molecules that inhibit enzyme targets,” Copeland said.
Accent is particularly focused on cancer, he noted, because that’s the area with the most extensive literature on the effects of RNA modification. Early in the company’s history, Accent researchers studied the human genome and identified hundreds of genes involved in RNA modification. They began systematically assessing the effects of knocking out those genes in cancer cells, looking for a promising anti-cancer result.
Those efforts produced about 1,500 candidate RNA-binding proteins, and from that the researchers picked out about a dozen that were attractive as precision oncology targets, Copeland said. A subset of those became discovery targets for small molecule inhibitors including the preclinical-stage ADAR1 program in head and neck cancer, non-small cell lung cancer, and tumors refractory to checkpoint inhibitors; a discovery-stage program targeting the RNA exonuclease XRN1 in the same set of cancers; and an undisclosed preclinical program in ovarian, triple-negative breast, small cell lung, and colorectal cancers. Accent also licensed a program targeting METTL3 to Ipsen in 2021 for R&D, manufacturing, and commercialization in acute myeloid leukemia.
However, DHX9, as Accent’s most advanced in-house program, made an attractive target, Copeland said, because it is “absolutely essential” for survival of cancers characterized by deficiencies such as DNA damage repair. At the same time, in normal cells, Accent has found that DHX9 is dispensable.
“We can define which cancers are likely to respond to DHX9 knockdown or inhibition with a small molecule, and in that way, identify patients that are most likely to benefit,” Copeland said. Accent has selected a lead drug candidate and begun investigational new drug application-enabling studies with the intention of submitting an application in the second half of 2024, which if cleared by the FDA will allow it to take the agent into clinical trials.
In preclinical studies, targeting DHX9 was lethal to cancer cells, and in mice, reduction of DHX9 expression did not have any adverse effects on body weight, blood biochemistry, and histology of various tissues compared to control mice. In data presented at the American Association of Cancer Researchers annual meeting earlier this year, Accent showed that inhibitors of DHX9 had anti-proliferative activity in MSI colorectal cancer cells with defective mismatch repair and that oral dosing of mice with the DHX9 inhibitor ATX968 resulted in robust and durable tumor regression correlating with intra-tumoral expression of the mRNA circBRIP1, a potential biomarker for clinical studies.
2023년 AACR Meeting에서 DHX9 Inhibitor 중 하나인 ATX968에 대해 발표를 했는데 mRNA circBRIP1 PD 에 Dose-Dependent Efficacy가 있슴을 보고했습니다.
Copeland said it is likely that Accent’s initial clinical studies for DHX9 will focus on MSI-high colorectal cancer, an indication well-supported by preclinical data. However, in the course of IND-enabling studies, Accent researchers will seek to understand the full spectrum of cancers that may be treatable with a DHX9 inhibitor before committing to a plan for clinical trials.
“We’re going to make our decision on the specifics of the clinical trial design probably in the first half of 2024,” Copeland said. In considering how to position such a drug on the market, Copeland added that the company would most likely establish activity of the drug in a relapsed or refractory patient population then attempt to build a case for its use in earlier lines of therapy. But, ultimately, those expectations could change depending on data collected from clinical studies. Biomarkers for patient stratification will also be an integral part of clinical trials for a DHX9 inhibitor, Copeland noted.
In addition to its internally run drug development efforts, Accent has also been active in pursuing partnerships for its pipeline programs. In June 2020, Accent inked a deal with AstraZeneca to discover, develop, and commercialize cancer therapies targeting RNA-modifying proteins. Accent received an upfront payment of $55 million from AstraZeneca in that deal and is eligible to receive up to $1.1 billion in milestone payments, option fees, and tiered royalties based on net sales.
Thus far, Accent has not actively pursued a partnership for the DHX9 program. However, following the data presentation at AACR, Copeland said, “That talk really created a lot of interest on the part of large pharma to engage with Accent. We’ve had many different conversations, and we are open to further conversations.”
이렇듯 프로그램들의 발전에 고무된 BMS, Johnson & Johnson 등이 참여하여 Series C $75 Million을 할 수 있었습니다. DHX9과 새롭게 공개한 KIF19A 약물들을 올해 연말까지 IND Filing을 하고 2025년에 임상1상을 할 예정이라고 공개했습니다.
Accent Therapeutics has reeled in investments from two more pharmaceutical companies as part of a $75M series C, the latest validation for the RNA drug developer.
Bristol Myers Squibb and Johnson & Johnson’s investment arm, JJDC, participated in the funding round that was announced Tuesday, which was led by the recently launched Mirae Asset Capital Life Science. The two pharmas join AbbVie’s venture arm which had previously participated in the biotech’s series B.
Accent CEO Shakti Narayan, Ph.D., said the money will help drive further development of the company’s two lead small molecules, targeting DHX9 and KIF18A, respectively. Accent’s plan is to formally ask regulators to enter human trials for both programs before the end of the year and put each in phase 1 studies by early 2025.
The three pharma backers come in addition to Accent’s existing research and licensing collaboration with AstraZeneca, announced in 2020. That gave the British pharma the option to a preclinical program that Accent would develop through phase 1 studies, plus worldwide licensing rights to two additional preclinical assets should AstraZeneca want more. Narayan wouldn’t specify which programs in the pipeline, if any, were assigned to AstraZeneca, saying only that the two lead programs were wholly owned.
All told, Narayan says the attention shows that Big Pharma “is excited about our story [and] is excited about the programs we’re driving forward.”
“Together with our collaboration with AstraZeneca, there’s just a great sense of endorsement and engagement from Big Pharma around our strategy and our vision,” he said.
The DHX9 asset has the potential to be first-in-class, but the KIF18A program is lagging slightly behind Volastra Therapeutics, which has two such assets in the clinic, one of which the biotech snagged from Amgen. Naveen Krishnan, M.D., managing director of Mirae Asset Capital’s new life science fund and incoming Accent board member, said Accent could gain from Volastra’s learnings.
KIF18A Program은 Volastra Therapeutics가 Sovilnesib과 VLS-1488의 두 프로그램이 이미 임상1상을 진행 중입니다. 이 데이타를 통해 Accent Therapeutics도 향후 이 프로그램의 향방을 결정할 수 있을 것 같습니다.
“Accent had a unique approach for how they’re pursuing their KIF18 asset that to me, really focuses on widening that therapeutic index,” Krishnan said. “And I think there’s certainly room for more than one player, particularly in KIF18A.”
Accent marks the first public investment for Kirshnan and Mirae’s new life science arm, which was unveiled last week with $50 million to spend. Krishnan, who took the reins after more than two years at Leaps by Bayer, expects the fund to make up to eight investments this year. He and the firm are already finalizing plans to participate in another unnamed biotech’s financing.
오늘 현재 Accent Therapeutics의 파이프라인은 아래와 같습니다. DHX9이 가장 앞선 상태이고 KIF18A가 그 뒤를 따르고 있습니다. RNA-Modifying Proteins를 표적하는 Accent의 전략과 RNA-binding Small Molecule을 개발하는 Expansion Therapeutics 등의 전략 중 어떤 회사들이 웃을지 궁금합니다.
 Geron’s The First Telomerase Oligonucleotide Inhibitor, Imeltestat, Near FDA Approval")



 GRO Biosciences: Genomically Recorded Organisms")





 Pearl Bio: Genomically Recorded Organisms with Synthetic Codons")


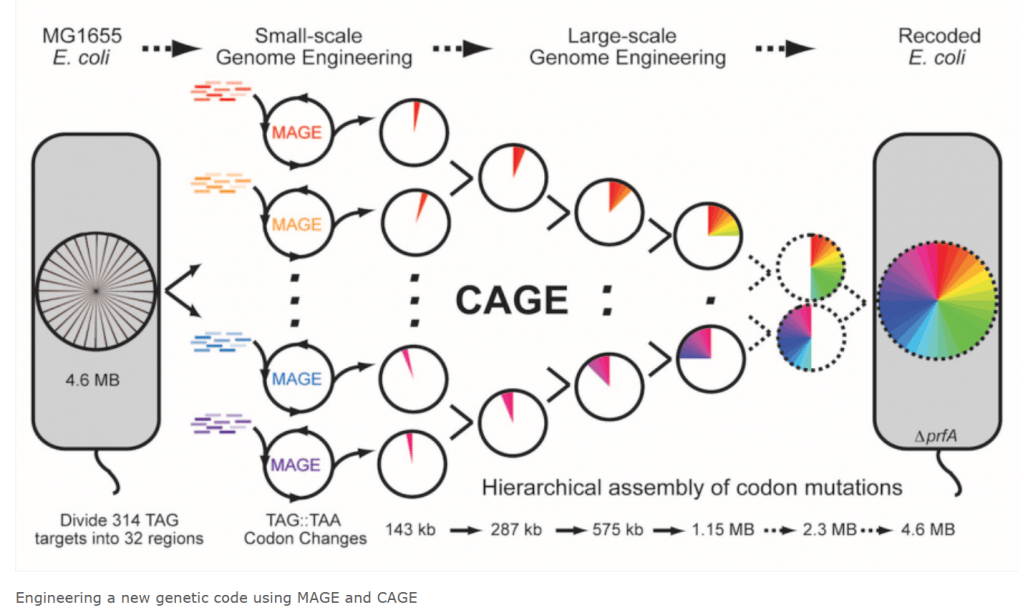
 Atlas Ventures: Option-to-Buy M&A Model – Nimbus Discovery & IFM Therapeutics")

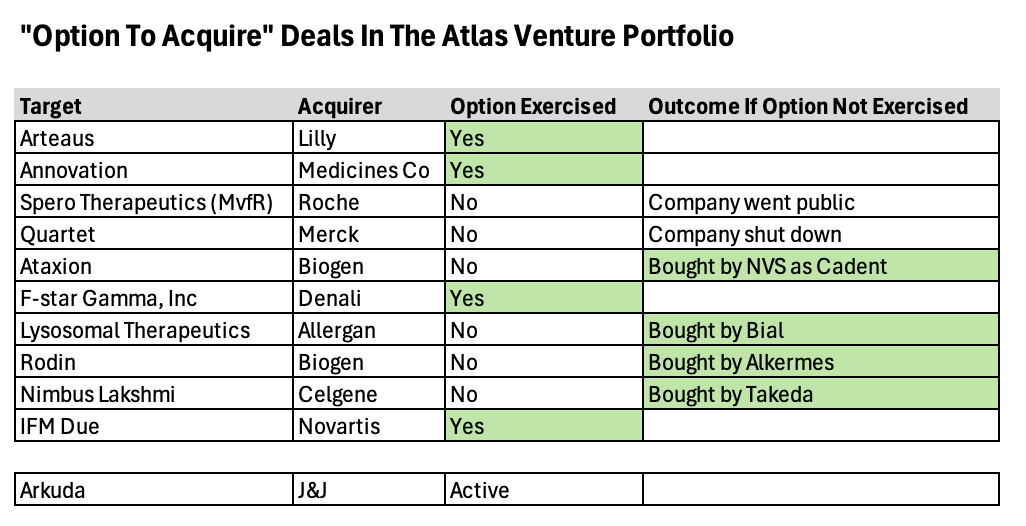



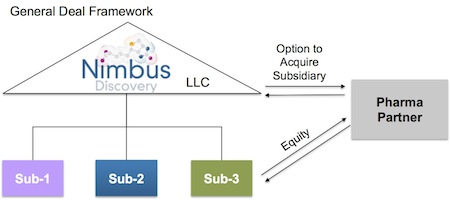



 편정현 헤드헌터 – 자리잡자")

 VLP Therapeutics: Self-Amplifying mRNA in Virus-Like Particle")



 Elie Dolgin, Ph.D. – Science Journalist")

 Calithera Biosciences의 실패에 대한 소고")






 Kenai Therapeutics: Allogeneic iPSC-Induced Neurology Cell Therapy")

 Accent Therapeutics: Small Molecules Targeting RNA-Modifying Protein")