현재 한국의 암환자들이 CAR-T치료제 치료를 받으려면 해외로 시료가 나가고 Autologous CAR-T 치료제가 만들어져서 QA/QC를 모두 통과하고 국내로 돌아와 환자가 다시 맞는데까지 너무 많은 시간이 소요됩니다. 이번에 한국 바이오텍이 자체개발한 CAR-T치료제로 한국의 환자들을 직접 치료할 수 있는 좋은 결과가 나온 것으로 생각해서 이에 대해 글을 남기려고 합니다.
큐로셀 (Curocell)은 2016년 당시 차바이오텍에 근무하던 김건수 대표가 KAIST 생명과학과 김찬혁 교수 (현재 서울대학교 교) 와 이화여대 심현보 교수와 함께 CAR-T, CAR-NK 세포치료제 개발을 위해 12월에 공동창업하게 됩니다. 본래 이 세사람은 서로 모르는 사이였는데 오름테라퓨틱스 이승주 대표의 소개로 의기투합하게 되었다고 합니다.
2017년에 인터베스트와 미래에셋캐피털로 부터 20억원의 시리즈A를 하고 2019년에 150억원의 시리즈B를 하게 됩니다. 스틱벤처스가 새롭게 참여했습니다. 이 자금으로 2020년 3월에 삼성서울병원 미래의학관 내에 GMP공장을 완공했고 2021년 2월에 식품의약품안전처로 부터 IND를 승인받아 임상1/2상을 시작했습니다. 2022년 1월에는 St. Jude Hospital로 부터 Lentivirus vector 기술도입을 했고 2023년 4월에는 신규 사옥(연구소, 본사) 및 상업용 GMP 공장을 완공(과학벨트 둔곡신동 거점지구)했습니다. 2023년 11월에 기술특례상장으로 코스닥 상장을 했고 최근 anti-CD19 CAR-T 치료제 안발셀 (Anbal-cel, Anbalcabtagene autoleucel)의 긍정적인 임상2상 데이타를 발표했고 식약처의 승인을 신청한 상태입니다.
PD-1과 TIGIT을 동시에 downregulation했을 때 CD19 CAR-T의 효능이 증대됨을 아래에서 보여주고 있습니다.
19PBBz (PD-1 downregulation) 19TBBz (TIGIT downregulation)에 비해 19PTBBz (PD-1/TIGIT Dual downregulation)이 CAR-T 치료의 지속성이 높은 것을 보여줍니다.
19BBz-nt에 비해 19PTBBz-nt (PD-1/TIGIT Dual downregulation)에서도 백혈구의 GM-CSF level이 높게 나온 것으로 볼 때 CAR-T치료제의 독성인 CRS (Cytokine Release Syndrome)과 ICANS (Immune effector Cell-Associated Neurotoxicity Syndrome)이 적을 것으로 기대할 수 있었습니다. (G)
Subcutaneous Raji-PD-L1 Lymphoma in vivo model에서 19PTBBz-nt (PD-1/TIGIT Dual downregulation)와 19BBz-nt의 in vivo efficacy를 보았을 때, 19PTBBz-nt (PD-1/TIGIT Dual downregulation)가 19BBz-nt 보다 모든 dose level에서 개선된 항암효과를 보였습니다.
삼성의료원 김원석 교수팀은 2022년에 Anbal-cel의 r/r large B-cell lymphoma (r/r LBCL)에서의 임상1/2 상 결과를 발표했습니다.
CAR-T 치료 전 3일간 Lymphodepletion with cyclophosphamide (500mg/m2) and fludarabine (30mg/m2) 을 수행했습니다. 그리고 세가지 Dose에서 한차례씩 IV 주사를 실시했습니다. 총 9명의 환자에게 시행했고 이 중 한분은 중환자실에 입원 (Grade 3), 5명 (56%) 은 CRS (Cytokine Release Syndrome) 독성을 보였습니다. 이 정도 독성은 CAR-T치료제에서 일반적인 것입니다.
9명 중 7명 (78%)이 CR (완전관해)를 보였는데 가장 낮은 용량이 3명, 중간과 최고 용량이 각각 2명이었습니다. 이 결과를 바탕으로 2022년 3월에 임상2상을 시작한다고 발표했습니다.
Background:Anbal-cel is a novel 2nd generation autologous CD19 CAR-T cell therapy which has been knock-downed for PD-1 and TIGIT using OVIS platform. Anbal-cel demonstrated the eradication of CD19 positive tumor cells in vitro and in vivo better than conventional CD19 CAR-T cells. The knock-down of PD-1 and TIGIT at CD19 CAR-T cells exerts the superior T-cell functionality by delaying the exhaustion of CAR-T cells.
Methods: This phase 1 dose escalation part (NCT04836507) was evaluated the safety and preliminary efficacy in patients with r/r LBCL. Anbal-cel was manufactured at GMP facility with fresh leukapheresis product. Patient was administered as a single intravenous dose at dose level 1 (DL1) (2×105 cells/kg), DL2 (7×105 cells/kg) or DL3 (2×106 cells/kg). Lymphodepletion with cyclophosphamide (500mg/m2) and fludarabine (30mg/m2) was performed for 3 days prior to Anbal-cel infusion.
Results: As of Jan 17 2022, 9 patients with r/r DLBCL were infused with Anbal-cel; 4pts at DL1, 3pts at DL2 and 2pts at DL2. Median age was 54 (range 26-71); all patients received 2 or more prior lines of therapy and 44% (4/9) received ≥4 prior line of treatment before the study. 78% (7/9) patients were refractory to their last treatment. 67% (6/9) of patients were at IPI 3-4 and 44% (4/9) of patients had bulky disease. No patient experienced DLT during the study. Of the 9 patients, 5 (56%) experienced CRS; 4 (44%) were grade 1 or 2 and one patient experienced grade 3 CRS. Median time to onset of CRS was 7 days (range, 1-16) with median duration of 4 days (range, 0-18). One patient dosed at DL3 experienced grade 2 ICANS, time to onset of ICANS was 7 days and lasted for 13 days. This patient had prior CNS involvement history before the study. Most commonly reported grade 3/4 AEs were neutrophil count decrease (6/9, 67%), anemia (5/9, 56%), thrombocytopenia (2/9, 22%), platelet count decrease (2/9, 22%) and no infection was reported. Complete response rate (CRR) was 78% and complete responses were observed at the lowest dose level and from patients expressing less than 10% CD19 at IHC; 3 complete responses (CR) at DL1, 2 CRs at DL2 & DL3 respectively. Dose-dependent CRC01 expansion was observed; median Tmax was 15.4, 15.8, 11.0 days at DL1, DL2 & DL3 each; median Cmax was 18,003, 30,103, 53,688 copies/ug gDNA at DL1, DL2 & DL3 each; median AUC0-28day was 679,125, 1,110,108, 2,852,235 copies/ug gDNA at DL1, DL2 & DL3 respectively.
Conclusions:Anbal-cel demonstrated promising efficacy and tolerable safety profile in this dose escalation study. Based on this phase 1 study, phase 2 patient enrollment will be commenced in Mar 2022 to evaluate the response rate, duration of response of CRC01 as well as safety. In addition, various biomarker studies are planned to investigate the differential mode of action of Anbal-cel during phase 2 study. Clinical trial information: NCT04836507.
삼성의료원 김석원 교수팀은 2023년에 임상2상 중간결과를 발표했습니다. 임상1상에서 최고용량이었던 2×106 cells/kg으로 r/r LBCL환자들에 대해 Biomarker analysis를 한 것입니다. 완전관해를 받지 못한 환자들의 경우에는 PD-1, TIGIT 등의 관문유전자 레벨이 상승한 것을 관찰해서 완전관해와 메카니즘 적으로 PD-1과 TIGIT 레벨이 중요한 영향을 끼침을 발표했습니다.
Background: Anbalcabtagene autoleucel is a novel anti-CD-19 CAR-T therapy that has shown promising clinical results, with a complete response (CR) rate of 73.7% in relapsed or refractory LBCL patients at interim analysis. This biomarker analysis aims to understand the mechanism associated with the treatment outcome. Methods: Patients with relapsed or refractory LBCL were enrolled to receive Anbal-cel at a dose of 2×106 cells/kg. Tumor biopsies were performed at baseline to assess the expression levels of CD19, PD-L1, and CD112/CD155. Peripheral blood mononuclear cell (PBMC) samples for immune phenotyping and serum sampled for ctDNA analysis were collected at day 0, 14, 28, and 3-month intervals thereafter until progression. Results: PD-1 and TIGIT were highly expressed on baseline PBMC. PD-1 was expressed more on CD8+ CAR+ T-cells whereas TIGIT on CD4+ CAR+ T-cells. PD-1, LAG-3 and TIM-3 expressions were significantly increased from D14 at non-CR group. TIGIT expression was significantly increased at D28 at non-CR group. The non-CR group demonstrated the significantly increased level of IL-4+/ IL-5+ Th2 type CAR+ T-cells whereas GATA+Th2/Tc2 type CAR+ T-cells were significantly lower in non-CR group. Additionally, CD57+CD27-CAR+ T-cells, a marker of terminal differentiation/ functional senescence, were significantly increased in the non-CR group from D14. CD226+ CAR+ T-cells were also significantly lower in the non-CR group. CD45RA+ CAR+ TEFF/EMRA cells postinfusion were significantly lower in the non-CR group and CD38+HLA-DR+ CAR+ T-cells, a marker of susceptibility of cell death, were significantly higher in the non-CR group. The activation of CAR+ T-cells measured by ICOS, 4-1BB, CD226 and CD94 was not different between the two groups. The most frequently detected mutations were related to cell cycling, epigenetic regulation, immune escape, apoptosis etc. Changes in ctDNA concentration were reversely correlated with treatment outcome.
Conclusions: Anbal-cel’s unique immunologic and genetic changes demonstrated potential to correlate with treatment outcomes and warrant to confirm with more data. Clinical trial identification: CRC01-01 (NCT04836507). Legal entity responsible for the study: Curocell Inc. Funding: Curocell Inc. Disclosure: Y. Koh: Financial Interests, Institutional, Advisory Board: Curocell. J.R. Kim: Financial Interests, Personal, Member: Curocell. All other authors have declared no conflicts of interest.
그리고 최근 73명의 임상시험 결과 CRR (완전관해율)은 67%였고 ORR (객관적 반응률)은 75%였다고 발표했습니다. 이 결과가 맞다면 아주 좋은 결과입니다. 물론 미국이나 유럽 임상에 비해 인원수가 적은 문제는 있지만 국내 r/r LBCL 환자수를 보았을 때는 식약처에 승인요청을 할만한 결과를 얻은 것으로 볼 수 있을 것 같습니다. 금년 하반기에 식약처에 승인요청을 할 예정이라고 공시했습니다.
■ 거대B세포림프종(LBCL) 환자 대상 임상 2상시험에서 완전관해 67.1% 확인 ■ 차세대 기술이 적용된 최초의 국내 개발 CAR-T 치료제로 연내 신약허가 신청 예정
큐로셀(대표이사 김건수)이 재발성, 불응성 거대B세포림프종(LBCL) 환자를 위한 차세대 CAR-T 치료제 ‘안발셀(Anbal-cel)’의 임상 2상시험 톱라인 데이터를 수령했다고 6일 공시했다.
임상2상 최종 데이터 분석 결과 임상시험 유효성 분석 대상자 73명 중 안발셀 투여 후 암세포가 모두 사라진 완전관해에 도달한 비율(CRR)은 67.1%였다. 이는 글로벌 시장에 출시된 CAR-T 치료제들의 기존 임상시험 결과와 비교해 가장 우수한 완전관해율이다. 또한 일차 평가변수인 객관적반응률(ORR, 전체 환자에서 약물의 객관적 반응이 나타난 환자 비율)은 75.3%이었다. 최종 결과는 임상시험 설계 당시 가정했던 통계적 유의성을 확보했다.
이번 결과는 2개 차수 이상의 치료에 재발 또는 불응하는 거대B세포림프종(LBCL) 환자를 대상으로 안발셀을 단회 투여한 후 안전성 및 유효성을 평가하는 공개, 다기관, 단일군 임상시험을 통해 얻어진 것이다. 해당 임상은 2022년 3월부터 2023년 10월까지 만 22세~85세 성인남녀 79명을 대상으로 삼성서울병원 등 6개 기관에서 수행한 임상 2상으로 국내 최초의 CAR-T 치료제 임상시험이다.
큐로셀은 이번 임상 결과를 토대로 올해 하반기 국내 신약허가를 신청할 예정이다. 안발셀의 신약허가 획득 시, 우리나라는 미국, 중국, 인도에 이어 자체적으로 CAR-T 치료제를 개발한 네 번째 국가가 될 전망이다.
큐로셀은 국내 최대 규모(1만 636m2)이자 글로벌 수준의 CAR-T 치료제 전용 상업용 GMP를 보유한만큼 신약허가 획득 후 국내 제조를 통한 공급을 진행할 계획이다.
큐로셀 김건수 대표는 “지난 3년간 매진했던 임상시험을 성공적으로 마무리해서 매우 기쁘고 관련한 모든 분들께 감사 인사를 드리고 싶다. 이번 최종 결과에서 안발셀의 높은 경쟁력을 확인한 만큼 향후 신약허가와 출시가 빠르게 진행되도록 최선을 다할 것”이라고 밝혔다.
회사의 홈페이지에 개재한 파이프라인은 다음과 같습니다. Anbal-cel은 현재 ALL (급성림프구성백혈병) 환자에 대해 임상1상을 진행 중에 있습니다.
큐로셀이 시리즈B로 150억원 규모의 투자자금을 유치했다고 23일 밝혔다. 기존 투자기관으로 인터베스트, 미래에셋캐피털이 후속 투자를 했고, 타임폴리오자산운용, 스틱벤처스가 새롭게 참여했다. 큐로셀은 2년전에 시리즈 A로 20억원을 투자받은 것을 포함해 총 170억원을 확보하게 됐다. 이번 자금유치를 통해 큐로셀은 임상 진입을 위한 연구개발 가속화하고 임상의약품 제조용 GMP시설 구축에 집중할 계획이다. 큐로셀은 제약업계에 다년간의 경험을 가진 김건수 대표와 T세포 치료제 전문가 KAIST 김찬혁 교수와 항체 전문가 이화여대 심현보 교수가 함께 창업한 국내 최초의 CAR-T 치료제 전문회사다. 큐로셀은 기존 CAR-T치료제의 한계를 극복하는 차세대 CAR-T 기술 개발에 포커스한다. 핵심 기술로 암세포를 인식할 수 있도록 CAR발현과 동시에 면역관문수용체를 통한 면역억제현삼을 극복할 수 있는 CAR-T 기술을 이용해 혈액암, 나아가 고형암 치료에 도전하고 있다. 최근 큐로셀은 2020년 차세대 CD19 CAR-T의 임상1상 진입과 신규 프로젝트 발굴을 위하여 삼성서울병원과 업무협업계약을 체결했다. 회사는 기초연구, 임상연구 및 임상의약품생산 등 면역항암세포치로제 개발 관련 전분야에 걸쳐 협업체계를 구축했다. 김건수 대표는 “큐로셀의 가치를 믿어 주신 투자자 분들께 감사드리며 삼성서울병원과의 업무협약에 이어 시리즈B 투자유치로 세계적으로 뒤처져 있는 국내 CAR-T 분야에서 빠른 임상 데이타 확보를 통해 기술력을 증명해 보이겠다”라고 밝혔다.
국내 최초 CAR-T 치료제 개발 전문기업인 큐로셀 (Curocell)이 440억원 규모의 시리즈C 투자유치에 성공했다. 큐로셀은 이에 앞서 지난해 1월 시리즈B 150억원 투자유치를 했으며, 이로써 지금까지 총 누적금액은 615억원이다. 큐로셀은 내년 상장을 계획하고 있다. 이번 시리즈는기존투자기관인 스틱벤처스, 에이티넘인베스트먼트가 후속투자를 진행하였고 DS자산운용, 서울투자파트너스, IMM인베스트먼트, 아주IB투자, 얼머스인베스트먼트, LB인베스트먼트, 유경PSG자산운용, 이앤벤처파트너스, JX파트너스, 컴퍼니케이파트너스, K2인베스트먼트파트너스, 쿼드자산운용, 하나벤처스 등 신규 투자기관이 대거 참여했다. 이번 투자금은 차세대 CD-19 CAR-T 치료제인 ‘CR101’의 연내 임상 개시와 후속 파이프라인 개발을 가속화하는데 투입된다. CR101은 기존의 킴리아, 예스카타 등 CD19 CAR-T 약물의 디자인에서 T세포 항암 활성을 억제할 수 있는 면역관문분자 TIGIT과 PD-1 발현을 낮춘 약물이다.
CAR-T 전문기업 큐로셀 (Curocell)이 공모가를 확정하고 내달 9일 상장할 예정이다. 큐로셀은 수요예측 결과를 반영한 최종 공모가로 희망 공모가밴드 (2만9800 ~ 3만3500원) 아래인 2만원으로 확정했다고 30일 공시했다. 이에 따라 총 공모규모는 320억원으로 줄어들었다. 확정공모가액 기준 시가총액은 약 2723억원이다. 큐로셀은 이번에 조달한 공모자금으로 현재 진행중인 차세대 CD19 CAR-T치료제 안발셀 (Anbal-cel, 성분명 안발캅타진 오토류셀)의 상업화와 함께 다발성골수증, T세포림프종, 고형암 등 새로운 파이프라인 개발에 투자할 계획이다. 김건수 큐로셀 대표는 “최근 주식시장의 분위기가 좋지 않았다. 특히 바이오기업들이 유독 더 어려움을 겪고 있는 상황에서 신규 상장 바이오 기업으로서는 올해 가장 큰 규모의 공모를 마무리하게 됐다”며 “어려운 시장 분위기에서도 큐로셀에 대한 믿음을 갖고 공모에 참여해 주신 투자자분들께 감사드린다”고 말했다. 이어 김대표는 “국내 최초로 CAR-T 치료제 개발을 시작하여 새로운 시장을 개척하고 있는 규로셀만의 기술력과 노하우를 기반으로 상장 후 글로벌 경쟁력을 갖춘 혁신적인 항암면역세포 치료제 전문 기업으로 도약하겠다”고 밝혔다. 큐로셀의 총 공모 주식 수는 160만주로 오는 31일부터 11월 1일까지 양일간 일반 투자자 청약을 진행한 뒤, 11월 9일 코스닥 시장에 상장할 예정이다. CAR-T치료제는 환자의 혈액에서 분리한 면역세포인 T세포를 유전적으로 조작해 암세포를 효과적으로 제거할 수 있도록 한 세포유전자치료제다. 림프종 등 혈액암 치료제에서 높은 반응률과 완전관해를 (CR rate)를 뵈며 전세계에서 주목받고 있다. 큐로셀은 국내 최초로 설립된 CAR-T 개발 전문기업으로 최근 안발셀의 거대비만성B세포림프종 (DLBCL) 대상 임상2상을 완료하고 내년 9월 신약허가 신청을 계획하고 있다. 큐로셀은 CAR-T 세포의 기능 저하 원인인 억제성 면역관문분자 PD-1, TIGIT 의 발현을 낮추는 OVIS 기술을 개발해 CAR-T의 기능을 강화하며 치료 효과를 높이겠다는 목표이다. 큐로셀은 국내를 비롯한 해외 25개국에 OVIS의 특허를 출원 중이며 현재 한국과 미국, 유럽, 일본 등에서 특허 등록이 완료됐다.
36명 벤처가 없던 길을 만들고 있다. 국내에선 누구도 가보지 않은 길이다. ‘기적의 항암제’라 불리는 CAR-T세포 치료제만을 개발하기 위해 설립된 바이오벤처 ‘큐로셀’ 얘기다. CAR-T세포 치료제는 말기 혈액암 환자 치료에서 80%까지 차도를 보이면서 2017년 미국 식품의약국(FDA)도 허가한 면역항암제다. 그러나 국내에선 아직 기술 축적이 이뤄지지 않아 개발되지 못하고 있다.
국내에서 누구도 엄두를 내지 못하는 가운데, 대덕 바이오벤처 큐로셀은 글로벌 제약사를 뛰어넘는 기술력을 입증하며 업계를 뒤흔들고 있다. 큐로셀은 T세포에 키메릭 항원 수용체(CAR)를 붙이는 유전자 조작을 넘어 면역관문 수용체 2종(PD1, TIGIT)을 제거하는 독보적 기술로 도전장을 던졌다. 글로벌 바이오 기업 대다수가 면역관문 수용체 하나(PD1)만을 제거한다. T세포 표면에는 면역관문 수용체가 여러 개 있는데, 여기에 암세포가 악수를 하듯 들러붙으면 T세포가 기능을 상실한다.
김건수 대표는 “유전자 조작을 통해 T세포에 CAR를 붙이는 건 어느 기업이나 할 수 있는 기술”이라면서 “대다수 기업들이 PD1 면역관문 수용체 하나만을 제거하지만, 내부에서 여러 조합을 테스트해본 결과 PD1과 TIGIT을 없앴을 때 CAR-T 치료 효능이 극대화된다”고 했다.
큐로셀의 독보적인 기술력을 알아본 건 국내 투자사들과 삼성서울병원이다. 큐로셀은 현재까지 총 615억원을 투자받았다. 삼성서울병원은 큐로셀이 개발하고 있는 CAR-T세포 치료제를 원내에서 생산할 수 있도록 내부 공간 495m2(150평)까지 내줬다. 그곳에 올해 초 CAR-T세포 치료제를 생산할 수 있는 GMP 시설이 완공됐다. 시운전과 자체 생산 테스트까지 끝난 상황으로 정부(식품의약품안전처) 임상 허가를 기다리고 있다.
◆ 개척 정신
김 대표는 어린 시절부터 틀에 박힌 일보단 하고 싶은 일을 찾아 나섰다고 한다. 새로운 일에 두려움 없이 나아가는 아버지 영향이 컸다. 그는 연세대 생명공학과에서 학·석사를 마치고 당시 한화석유화학중앙연구소, LG생명과학, 차바이오텍 등을 거치면서 CAR-NK세포에 관심을 두기 시작했다. 2015년 CAR-NK세포 치료제를 시작으로 CAR-T세포 치료제를 독학하던 시기 아버지가 암으로 세상을 떠났다. 이듬해 그는 운명처럼 면역항암제만을 전문으로 하는 바이오벤처를 설립해 이전에 없던 길을 나아가고 있다.
김 대표는 “암 환자에게 CAR-T세포 치료제는 마지막까지 해볼 수 있는 가능성 높은 치료”라면서 “암 환자에게 효능이 있다고 증명된 만큼 국내에도 반드시 있어야 하는 치료제라고 생각했다”고 말했다.
김 대표는 어려움을 묻는 질문에 “연구개발을 하고 결과를 기다리는 입장에서 미래에 대한 불확실성은 늘 있는 것이고 그걸 난관이라고 할 수는 없다”면서도 “국내에서 CAR-T는 모든 게 처음이기 때문에 물건을 사거나 원료를 주문 생산해야 하는 상황 하나하나가 난관이지만 극복해나가고 있다”고 했다.
◆ “목마른 사람이 우물 판다”
김 대표는 연구원 생활을 본격 시작한 2000년부터 창업 전까지 커리어 15년을 연구자와 기획자로 절반을 살았다. 그는 이 시기 연구에 대한 전문성도 키웠지만, 기획자로서 커뮤니케이션 내공을 쌓았다고 한다. 그는 “기획 일을 하면서 얻은 건 사람들 간 이해관계 조정이나 연구 분야에 대한 존중이 필요하다는 점”이라고 설명했다.
그는 이런 내공을 통해 큐로셀을 CAR-T 최고 전문가들로 채웠다. 이승주 오름테라퓨틱 대표를 통해 김찬혁 KAIST 생명과학과 교수를 소개 받아 사업을 고도화시켰고, 항체 전문가인 심현보 이화여대 교수도 영입했다. LG화학에서 바이오시밀러 개발을 책임지던 김형철 현 큐로셀 상무를 데리고 오기도 했다. 기획 일을 하면서 커뮤니케이션 내공, 사람 만나는 일에 거리낌 없는 김 대표의 능력이 진가를 발휘한 것이다.
김 대표는 “혹자는 아무것도 없는 상황에서 어떻게 창업할 수 있느냐고 묻지만, 어느 기업보다 창업 멤버들이 훌륭하고 아이템도 세계적으로 유망하기 때문에 여러 면에서 불안할 요소가 없는 비즈니스”라고 자신감을 드러냈다.
삼성서울병원 GMP 시설을 구축하는 과정에도 김 대표는 서울대병원과 삼성서울병원 등을 직접 찾아 나섰다. 당시 CAR-T세포 치료제에 대한 회의적인 시각도 있었지만, 좌절하지 않고 그간 쌓아 온 내공과 기술력으로 삼성서울병원 내부에 GMP 시설을 구축했다.
김 대표는 “목마른 사람이 우물을 파는 것”이라며 “사람 안 만나고 일할 수 없고, 일을 하려면 무엇이든 부딪쳐봐야 한다”고 했다. 이어 “움직이지 않으면 누군가 대신해줄 수도 없다”며 “그렇기 때문에 핵심 인력을 모으고 국내에서 처음으로 하는 CAR-T세포 치료제 생산을 위해 GMP 시설 구축이 필요했던 것”이라고 설명했다.
그는 “이 기반 위에서 림프종 임상 시작이 목표이고 백혈병, 다발성골수종까지 범위를 넓혀가겠다”며 “혈액암뿐만 아니라 고형암에서도 CAR-T를 쓸 수 있도록 기술을 개발하고 새로운 길을 나아가는 것이 목표”라고 강조했다.
◆ CAR-T 기술과 큐로셀 차별성
환자 혈액 채취→ 백혈구에서 T세포(면역세포) 분리 → 유전자 조작을 통해 T세포에 특정 암을 인식하는 유전자(CAR)를 삽입 → *T세포에 있는 면역 관문 수용체(PD1, TIGIT) 2종 제거 → T세포 배양 → 환자에게 투약.
*큐로셀은 면역세포(T세포)에 있는 면역 관문 수용체 2종(PD1, TIGIT)을 제거해 CAR-T세포를 만드는 기술이 독보적이다. 암세포는 T세포 표면 면역관문 수용체에 달라 붙어 T세포 면역 기능을 무력화 시킨다. 대다수 바이오 기업은 PD1이라는 면역 관문 수용체를 제거하는 것과 달리 큐로셀은 PD1과 TIGIT을 제거해 CAR-T 효능을 극대화하고 있다.
◆ 용어 설명
☞ 림프구(Lymphocyte)
림프구는 면역계를 구성하는 중심 세포다. 우리 몸 속에는 감염원으로부터 신체를 보호하는 면역계 세포 ‘백혈구’가 있다. 전체 백혈구 중 약 25% 정도를 차지하는 세포를 림프구라 일컫는다. 림프구는 적응 면역과 항원 특이성, 수용체의 다양성, 기억, 자기 비자기 구분이라는 특징을 지닌다. 림프구는 조혈모세포에서 만들어지며 성숙된 림프구는 기능에 따라 B세포와 T세포로 나눌 수 있다.
☞ T세포(T cell)
면역 반응에 관여하는 림프구는 B세포와 T세포가 있다. B세포는 체내에 침입한 세균·바이러스에 대항하는 항체를 만든다. T세포는 몸속에서 여러 기능을 하는데, 그중 면역에서 기억능력을 지녀 B세포에 정보를 제공해 항체 생성을 돕는다. 특히 T세포는 병원체에 감염된 세포를 직접 사멸시키는 능력을 지닌다. 이 뿐만 아니라 면역 활동을 적절히 조절하는 기능도 지닌다.
☞ CAR-T세포 치료제(Chimeric Antigen Receptor T cell Therapy)
환자 혈액에서 얻은 T세포와 암을 잘 인지하는 키메릭 항원 수용체(CAR)를 유전자 조작해 만든 세포 치료제다. 환자의 혈액에서 T세포를 추출한 뒤 바이러스 등을 이용해 암세포에 반응하는 수용체 DNA를 T세포에 주입하고 증식 시켜 몸속에 넣어주는 방식이다. 암세포와 수용체가 ‘열쇠와 자물쇠’처럼 결합하면 T세포는 암세포를 공격하는 원리다. CAR-T 세포 치료제는 정상 세포 손상을 줄이면서 효과적으로 암세포를 사멸할 수 있는 차세대 항암제다.
Eli Lilly와 Novo Nordisk의 Oral GLP-1R Agonist를 Type 2 Diabetes 치료제와 비만치료제로서 개발하는 경쟁이 매우 뜨겁습니다. 작년 6월에 열렸던 ADA (미국당뇨학회)에서 Novo Nordisk의 Oral Semaglutide는 15%(68주) 의 체중 감량을 보였고 Eli Lilly의 Orforlipron도 15% (36주)까지의 체중감량을 발표했습니다. 사실상 거의 동일한 결과라고 볼 수 있습니다. 실로 강대강 대결 양상입니다.
The potential advantages of a daily-pill version of popular GLP-1 drugs for Type 2 diabetes and obesity are obvious compared to the weekly injection routine most patients taking these drugs undergo. Last weekend at the American Diabetes Association (ADA) Scientific Sessions in San Diego, market leaders Novo Nordisk and Eli Lilly presented data that showed their investigative oral GLP-1 treatments are making progress.
A phase 3 study of Novo Nordisk’s high-dose oral semaglutide, OASIS 1, shows that obese patients averaged a weight loss of 15% after 68 weeks of treatment, with 34% seeing a 20% drop in their weight. The results are comparable to the weight reductions seen with Novo’s injected GLP-1 drugs.
Also at the ADA conference, Lilly presented results from a phase 2 trial of its weight-loss pill orforlipron, which produced average weight losses of between 9% and 15% depending on the dose provided over a 36-week period. Doses tested were 12 mg, 24 mg, 36 mg and 45 mg. Results were published Friday in The New England Journal of Medicine.
Eli Lilly가 임상2상을 진행 중인 Orforlipron (OWL833/LY350297)는 Roche의 일본 자회사인Chugai Pharmaceutical이 개발한 약물입니다.
Chugai 제약과 Eli Lilly 연구원은 2020년 PNAS 논문에 Orforlipron (OWL833/LY350297)의 개발에 대해 발표했습니다.
Cyno Molgus를 이용한 NHP Study 결과는 다음과 같습니다. Exenatide i.v. 주사제와 비교한 실험에서 대등한 결과를 보여주어서 Orforlipron (OWL833/LY350297) 경구투여제의 임상시험을 지지했습니다.
Eli Lilly & Co. is looking to pad out its diabetes portfolio by licensing an oral, non-peptidic GLP-1 receptor agonist from Chugai Pharmaceutical that the company describes as a “phase 1 ready” asset for the treatment of Type 2 disease.
In return for $50 million upfront, Lilly will receive worldwide development and commercialization rights to OWL833, with Chugai, a Tokyo-based member of the Roche Group, eligible for future milestone payments and royalties.
“We believe OWL833 can be a best-in-class oral non-peptide GLP-1 receptor agonist and that its value will be further enhanced through Lilly’s clinical development to contribute to people around the world who live with diabetes,” Yasushi Ito, M.D., Ph.D., Chugai’s executive VP and co-head of its Project & Lifecycle Management Unit, said in a statement.
Both companies said there would be no changes to their financial forecasts or guidances for 2018 as a result of the deal, and that OWL833 will “soon” enter phase 1 clinical development.
Lilly has been working to grow and broaden the technologies in its diabetes pipeline, as well as extend its returns with stronger forays into devices and real-world evidence generation.
In April, the company paid $63 million for Type 1 diabetes cell therapies from Sigilon Therapeutics, and signed on for an additional $410 million in possible milestone payments.
The Cambridge, Massachusetts-based Sigilon aims to induce pluripotent stem cells become insulin-producing beta cells, encapsulated using its Afibromer islet cell technology to protect the implanted cells from the immune system.
A few months prior, Lilly unveiled a device-driven strategy to weather pricing pressures, following its partnership with Dexcom to develop continuous glucose monitoring systems. Lilly has also been working on an automated, wearable insulin delivery device and smart pen injector at a small lab it launched in 2015.
2023년 New England Journal of Medicine에 Orforglipron의 임상2상 결과를 발표했습니다.
Non-peptide GLP-1R Agonist로 Pfizer의 Danuglipron이 twice-a-day oral GLP-1R Agonist 약물로 개발 중이었지만 최근에 독성문제로 인해 임상이 중단되었습니다. 대신에 once-a-day oral formualtion의 PK data를 통해 새로운 임상이 시작될 수 있슴을 얘기했습니다. Pfizer는 oral GLP-1 pill의 시장규모를 $30 Billion으로 예상하고 있습니다.
Pfizer’s drug, known as danuglipron, is a GLP-1 agonist like in-demand obesity treatments from Novo Nordisk and Eli Lilly. But unlike those drugs, which are injections, danuglipron is taken orally, an advantage in convenience that Pfizer hopes will help it break into the fast-growing market.
Pfizer didn’t break out adverse event rates by danuglipron dose tested, but said up to 73% of participants experienced nausea, up to 47% vomiting and up to 25% diarrhea. The side effects were generally mild, the company said.
“We believe an improved once-daily formulation of danuglipron could play an important role in the obesity treatment paradigm, and we will focus our efforts on gathering the data to understand its potential profile,” said Mikael Dolsten, Pfizer’s top scientist and head of R&D, in the company’s statement.
Results from the ongoing pharmacokinetic trial of the once-daily version will “inform a potential path forward,” Dolsten added.
Another setback could put Pfizer in the uncomfortable position of falling further behind in a fiercely competitive development race. Lilly, which recently won U.S. approval of its GLP-1 weight loss drug Zepbound, is also developing an oral obesity medicine called oforglipron that showed promise in a mid-stage trial earlier this year.
Pfizer’s CEO Albert Bourla has said he expects the market for GLP-1 drugs, which are also used to treat Type 2 diabetes, to eventually reach $90 billion in sales. He estimated oral versions could claim about one-third of that figure.
일본의 제약회사 중 하나인 Chugai Pharmaceutical Co. Ltd. (中外製薬株式会社)는 2002년부터 Roche의 계열사로 되어 있는 회사로 신약개발을 하는 회사입니다. Chugai가 최근 mRNA Display Platform을 통해서 Macrocyclic Peptides 를 통한 신약개발을 중점적으로 하고 있는데 그에 대해 얘기를 하려고 합니다.
We report a versatile and durable method for synthesizing highly N-alkylated drug-like cyclic peptides. This is the first reported method for synthesizing such peptides in parallel with a high success rate and acceptable purity that does not require optimizations for a particular sequence. We set up each reaction condition by overcoming the following issues: (1) diketopiperazine (DKP) formation, (2) insufficient peptide bond formation due to the steric hindrance of the N-alkylated amino acid, and (3) instability of highly N-alkylated peptides under acidic conditions. Using this newly established method, we successfully synthesized thousands of cyclic peptides to explore the scope of this modality in drug discovery. We here demonstrate the syntheses of a hundred representative examples, including our first clinical N-alkyl-rich cyclic peptide (LUNA18) that inhibits an intracellular tough target (RAS), in 31% total yield and 97% purity on average after 23 or 24 reaction steps.
최근에 Journal of American Chemical Society에 LUNA18의 신약개발에 대한 논문을 게재했습니다.
Cyclic peptides as a therapeutic modality are attracting a lot of attention due to their potential for oral absorption and accessibility to intracellular tough targets. Here, starting with a drug-like hit discovered using an mRNA display library, we describe a chemical optimization that led to the orally available clinical compound known as LUNA18, an 11-mer cyclic peptide inhibitor for the intracellular tough target RAS. The key findings are as follows: (i) two peptide side chains were identified that each increase RAS affinity over 10-fold; (ii) physico-chemical properties (PCP) including Clog P can be adjusted by side-chain modification to increase membrane permeability; (iii) restriction of cyclic peptide conformation works effectively to adjust PCP and improve bio-activity; (iv) cellular efficacy was observed in peptides with a permeability of around 0.4 × 10–6 cm/s or more in a Caco-2 permeability assay; and (v) while keeping the cyclic peptide’s main-chain conformation, we found one example where the RAS protein structure was changed dramatically through induced-fit to our peptide side chain. This study demonstrates how the chemical optimization of bio-active peptides can be achieved without scaffold hopping, much like the processes for small molecule drug discovery that are guided by Lipinski’s rule of five. Our approach provides a versatile new strategy for generating peptide drugs starting from drug-like hits.
Journal of Medicinal Chemistry 2022년 논문에서 N-alkyl Rich Drug-like peptide synthesis를 개발해서 한 연구원 당 1년에 500개의 Peptides를 만들 수 있도록 했습니다.
Solid-phase synthesizer로 만드는 방법입니다. Cyclization을 먼저 한 이후에 Protecting group deprotection을 합니다.
최근에 Chugai R&D Day에서 발표한 자료에도 LUNA18에 대한 내용이 있습니다.
Chugai 제약은 Small molecule과 Antibody 신약개발은 오랜 기간 내공이 있는데 최근에 “intracellular Transferring Peptides” 분야에 Positioning을 한다는 전략을 수행하는 중에 있습니다.
개념도를 보면 Antibody는 Membrane Receptor에 결합하는 방법이고 Small molecule의 특성을 가지면서 Antibody와 같은 선택성을 갖는 Mid-size molecule을 발견하는 것이 목표라는 것입니다.
Mid-Size Molecule 중 처음으로 임상에 진입한 물질이 LUNA18입니다. pan-mutant RAS Inhibitor로 개발을 한 것입니다.
9-11-mer Macrocyclic Peptides가 Druglike한 Metabolical Stability를 위해서는 반 이상이 N-alkylated 되어야 한다는 것이 중요한 핵심 중 하나입니다.
PURESystem으로 Unnatural Macrocyclic Peptides를 만들 수 있는 mRNA Display Platform을 이룰 수 있었다고 발표했습니다.
PURESystem에서 Cyclization Method가 Key Reaction이고 Cyclization 이후에 Desulfurization을 합니다.
그리고 PeptiDream의 FIT (Flexizyme-mediated Flexible In Vitro Translation)이 N-alkyl amino acid를 받아들일 수 있도록 하기 위해 변형을 주었습니다.
이렇게 다양한 Platform Technology를 이용했을 경우에 Hit Identification부터 Lead Optimization까지 약 3년의 시간이 걸렸습니다. 역시 Lead Optimization이 시간이 많이 걸립니다. Cell-based system에서 active molecule을 찾은 후 DMPK를 위한 Lead optimization을 나누고 있습니다.
이러한 노력의 결과로 얻어진 LUNA18은 KRAS-G12C, KRAS-G12D, KRAS-G12V mutants에 모두 active하지만 KRAS-WT에서는 전혀 효과가 없기 때문에 정상세포에는 독성이 적고 Oncology KRAS mutants에만 듣는 Precision Oncology Medicine으로 될 가능성이 높은 약물입니다. In vivo efficacy도 dose에 따라 암세포가 줄어드는 것을 확인했고 체중은 대신 일정하게 유지가 되었습니다.
2023년 12월에 발표한 Chugai R&D Meeting Presentation은 아래에 링크합니다.
Novartis의 신약개발 Director이면서 Science에서 “In the Pipeline”이라는 블로그를 쓰는 Derek Lowe가 최근에 Chugai가 개발한 LUNA18 (pan-KRAS Inhibitor)에 대해 글을 쓴 것이 있습니다. Derek의 관점은 Chugai 연구팀이 N-alkyl amino acid를 이미 mRNA Display와 Solid-phase synthesizer에 도입을 해서 5번만의 In vitro selection만으로 180 nM active molecule을 찾았다는 점을 아주 높이 평가하고 있습니다. (Big Pharma Library Screening으로도 이런 것은 얻을 수 없다는 것을 강조하면서).
Chugai Pharmaceutical 연구팀이 Cell-Permeable Orally Active Macrocyclic Peptides를 위한 Druglike Platform을 한층 Upgrade한 것은 확실한 것 같습니다. 이 논문을 본 다른 연구팀들이 또한번 업그레이드 시키겠죠. 이 분야의 발전이 큰 기대가 됩니다.
I’ve been meaning to blog about this paper from a large team at Chugai, looking at ways to make rather large cyclic peptide structures that can also still be drugs. The whole “peptides as drugs” topic has been a perennial here on the blog, and by that I mean “going back to 2002“, with updates along the way. Here’s a recent review on the subject (and there are plenty more out there!)
The reasons it’s such a focus in drug discovery come from both ends of the topic. On the one hand, an awful lot of protein functions in the cell are mediated by, well, other proteins, or peptide pieces thereof. Protein-protein interactions (PPIs) are so ubiquitous, and the proteome that we have have is so tuned up for them, that a great many of our small-molecule drugs are actually fitting into binding sites that are normally part of some protein-binding event. (There are of course binding sites that are evolved for small molecules, such as with the amine GPCRs, and drug discovery efforts have naturally pounded away at those over the years, fear not).
But at the same time that there are a lot of protein-protein sites to exploit, actually getting down to expoiting them is difficult if you try to do it with an actual peptide, as opposed to some small molecule that ends up acting as a peptidomimetic instead. That’s because proteins of all sorts are constantly being recycled and remodeled in living cells. There are all varieties of saw blades spinning constantly in the biochemical environment, protease and peptidase enzymes that are ready to start slicing proteins up into smaller pieces. Our endogenous proteins are adapted to this, generally by being compartmentalized away from things that would chew them up and by not displaying easily-cleaved sequences to the enzymes they’re most likely to encounter. Instead, these protein-processing events are managed in a vast and intricate landscape, with a good example being the coagulation cascade.
Even getting to the stage where all these enzymes can take a whack at your peptide drug is not so easy, thanks to the way the digestive system is set up. We do not schlork up proteins as whole species when we eat them – instead, everything gets broken down thoroughly by digestive enzyme into individual amino acids, dipeptides, and tripeptides. Those are the species that are actually absorbed, and our innards are very good at ripping a huge variety of proteins into such sawdust. Which is what will happen to your drug candidate unless you take great care to avoid it.
There are more strategies than I can count for trying to fix these problems, and they have been refined and extended for decades now in drug discovery. N-methylation, reverse-chirality residues, beta-amino acids, “retro-inverso” chains, cyclic peptides of many kinds from simple rings to complex knots. . .those are some of the classics, and that’s just the start of the topic. The Chugai paper linked above is a contribution to this field, and a key step they’re taking is to start with the right sort of screening library – one that’s already most of the way to drug-likeness.
That means (they say) cyclic peptides, in roughly the 11-amino-acid size range, with more lipophilic side chains than usual, and a prominent amount of N-alkylation already built in. They’re taking their cues from cyclosporine, which is a notably effective compound with far better membrane penetration and pharmacokinetic behavior than one might have predicted. (Medicinal chemists have been mining the behaviors of such naturally-occurring macrocycles for a long time now!) The hope is that good hits from such a collection can be optimized without doing too much violence to the overall conformation of the ring and its physiochemical properties. Indeed, checking a library of 8-to-12-amino-acid membered ring cyclic peptide compounds showed that the 11-AA-membered ones had notably better stability to metabolic enzymes, and this is surely No Accident, evolutionarily. Similarly, it looks like you would want at least 6 alkylated residues (cyclosporine has 7) and a cLogP of at least 12.9 (cyclosporine’s is 14.4). Note: that is indeed quite greasy. And you’d like to have no more than 3 hydroxyl groups and a maximum of one ionizable group hanging off the structures as well (and probably none at all). Dosing a range of such compounds in mice confirmed that they were on the right track.
That’s a good amount of work already, but the group went on to work up an expression system to turn out large numbers of variants in this area for a screening library. That’s a challenge, because you’re asking the cellular protein synthesis machinery to do a number of things it normally doesn’t: you need it to handle plenty of N-alkylated amino acids, and what’s worse, you need to have some of these show up one after the other in the chains. You need to make macrocycles without relying on labile groups like disulfides or thioesters. And you need to strip out all the amino acids with ionizable side chains. That involves lot of engineering at the aminoacyl tRNA stage, but they managed to get an mRNA display system working with these modifications (using the PURE system, which is done outside of living cells and is thus more amenable to all the necessary changes).
mRNA display can give you a tremendous number of possible products, and indeed, the team ran an experiment that was capable of generating different peptides in the ten-to-the-tenth (tens of billions) range, and deploying this in a search for a KRAS ligand. That’s just the sort of audacious target this sort of technology should be applied to! Recent years have seen progress in targeting mutant forms of that cancer protein, but wild-type KRAS is a major challenge for anyone. After five rounds of enrichment, panning away all the less-potent candidates, they narrowed down on a particular cyclic peptide that had 180 nM activity in blocking the interaction of KRAS and its partner SOS1. You can screen the entire collections of big pharma companies and not find a compound like that (they have!)
The authors present an X-ray structure of the complex, which generally ends all arguments, and go on to show optimization of the compound into an even better drug candidate. As they’d hoped, this was achieved without any major alterations to the core structure, but rather relying on changing some side-chain properties. No polar (hydrogen-bonding) groups were used in that process – as they say, “it was not necessary to use polar functional groups to optimize the structure of a hit that does not rely on polar functional groups for binding to its target protein.”
These extra hydrophobic groups actually improved the properties of the new compound, which had sub-nanomolar activity for the KRAS-SOS1 interaction. It has good PK properties (47% oral bioavailability in dogs, for example), and is now in Phase I human trials. Which is pretty damned impressive (here’s another detailed look at the compound’s development). Chugai’s dedication to getting this macrocyclic-peptide-screening platform off the ground is impressive as well, and I very much look forward to seeing what else that can make out of it. And what the rest of the industry can make of the ideas behind it!
Medical researchers have leveraged technology to create major breakthroughs in the past few decades, accelerating the understanding of diseases, and their causes and treatment. Our accumulating knowledge also has accelerated the ability to translate science into practical therapies, but there are still many challenges: while researchers seek the right drug compounds that can target and deliver treatment for specific diseases, traditional drug innovation models can be slow and come with high costs.
Japan’s rising biotech company, PeptiDream, is tackling these issues, deploying a unique proprietary drug development technology and an innovative business model that will further research on and development and manufacture of peptides to deliver new medical therapies. “We really want to be a drug discovery engine,” says CEO Patrick Reid. Until recently, most advances in drug delivery have focused on small-molecule and large-molecule drugs, also known as antibodies. But now macrocyclic peptides are emerging as an important new avenue.
How are peptides different? Both small- and large- molecule drugs come with advantages – and limitations. The small molecule drugs are chemically synthesized in a lab and taken as a pill or capsule, so the active ingredient is easily absorbed into the bloodstream. Because they are small, molecules can penetrate cell membranes, making these drugs highly effective. But they can be unstable and they break down in the body, creating unwanted side effects. Formulating these drugs to take on specific new targets also can be slow and expensive.
Protein-based therapeutics (large-molecule drugs) are made by using living cells. They typically are not pills, but instead must be injected or infused. These large- molecule therapies, unlike the smaller-molecule drugs, cannot penetrate cells. But these drugs are easier to design for specific targets — typically a cell-surface receptor on the outside of the cell. However, these therapies cannot reach all required targets, and they can stay in the body too long causing side effects.
Enter peptides, compounds that consist of amino acids linked together and can be synthesized in the lab. Pioneering research by Suga Hiroaki, PeptiDream co-founder and professor at the University of Tokyo, established a way to ensure that a new kind of peptide compound can remain stable in the body and find a range of therapeutic targets with high specificity. They can also be broken down by and cleared from the body with greater specificity, making them an important new development in pharmaceuticals.
And these macrocyclic peptides can be combined, using a much larger set of amino acids than occur in nature – giving researchers the ability to experiment with many more combinations. PeptiDream’s Peptide Discovery Platform System (PDPS) is a proprietary technology that allows drug researchers to make trillions of peptide libraries. Reid describes PeptiDream as “platform company” that enables his researchers and others to make the process of discovering “hits” — the starting point for developing drugs — more efficient.
PeptiDream의 PDPS (Peptide Discovery Platform Systems)은 아래와 같습니다. 3천개 이상의 Amino acid로 다양한 Macrocyclic Peptides를 만들고 mRNA Display로 cDNAs를 만듭니다. 이 과정에서 1조개 이상의 Peptide library를 만들고 이 Library를 가지고 Target Protein과 가장 높은 Binding Affinity를 가지면서 높은 Selectivity를 갖는 Macrocyclic Peptides를 발굴합니다. 이 과정을 반복하면서 계속 Optimization한다는 개념입니다.
“We are not simply developing a single drug and trying to bring that all the way to approval; we are championing and developing an entirely new class of molecules,” Reid says. The platform has created an unusual set of collaborations for PeptiDream, whose drug discovery partnerships have included Merck, Bayer, Genentech and Novartis. This collaborative network has accelerated peptides development, Reid says, creating a large wave of compounds that should move into the clinic in the next few years.
“Our network of partners has allowed PeptiDream to function as company ten to twenty times its actual size,” he says. “With more than 100 discovery programs in parallel across a wide range of diseases, targets and administration routes, we are expanding the knowledge, understanding and appreciation of these molecules in therapeutics and diagnostics and more.”
It also was crucial that PeptiDream, as a startup, was able to focus on developing the platform for peptide drug discovery, something large pharmaceuticals had not done because of the cost and the long, uncertain time horizon. Japan embraced PeptiDream, initially as a largely bootstrapped company, and then when it went public in 2013, Reid says. “In the U.S. and Europe, we probably would have been pressured to borrow funds in order to grow faster,” he says. “Many companies in the U.S. with internal pipelines fail due to time and pressure constraints. They burn a lot of money very quickly.”
PeptiDream is now a $7 billion company and is also a founding investor in a contract manufacturing company, PeptiStar. Collaboration with other companies is crucial, says PeptiStar CEO Kameyama Yutaka, as new ecosystems for research, manufacturing and supply of peptide drugs are developed. In fact, PeptiDream, together with other co-founding investors Shionogi and Sekisui Chemical, has attracted additional ten investors as active R&D collaborators.
In order to accelerate the practical application and market creation of peptide therapeutics as next-generation drugs beyond biopharmaceuticals, the Japanese government also supports PeptiStar, providing 9 billion-yen (about $83 million) grant as part of the government’s program Cyclic Innovation for Clinical Empowerment, under the National Healthcare Policy. The money will allow PeptiStar, established in 2017, to become a leader in both scientific and business process innovation, says Kameyama.
“The current capacity of peptide manufacturing is limited, and it could be a big bottleneck of peptide medicine developments,” he says. “This quick fundraising will accelerate the development and commercialization of our ability to prepare the peptide compounds. And the support they have given us will also encourage many other partners in the important development of peptides.”
“Peptides have not been around very long, and as with any new technology, there is room for improvement, including costs,” Kameyama says. “We want to make production cheaper and higher quality, and collaboration is a competitive advantage. If a company established its own manufacturing facility, it would take time and money. But with a joint venture like ours, the cost and the sharing of technology and knowledge are very different.”
Both Reid and Kameyama credit the Japanese research and business ecosystem with their success. Professor Suga’s breakthrough work is just one spinoff of innovation coming from Japanese universities, where a pool of highly skilled research workers has developed. Japan’s challenge to create peptide drug market continues.
Peptidream이 2006년 7월에 UTEC과 설립한 이후에 2년간은 Stealth-mode로 회사의 PDPS (Peptide Discovery Platform Systems)을 만들고 IP를 확보하기 위해 노력을 했고 2008년에 펀딩을 받았고 2013년에 $52 Million로 동경 Mothers 상장을 했습니다.
PeptiDream Inc. (Tokyo, Japan) raised Y5.3 billion ($52.4 million) through the sale of 2.1 million shares at Y2,500 in an IPO on the Tokyo Stock Exchange’s market of the high-growth and emerging stocks (Mothers). The share figure includes the sale of 405,000 shares in an overallotment. The company’s founders are selling an additional 1 million shares in the IPO. The Y2,500 price, which is the upper end of PeptiDream’s proposed Y1,920-Y2,500 range, values the company at Y33.1 billion ($329.7 million). Mizuho is lead underwriter. The shares are slated to start trading June 11. …
“People thought I was crazy, because it’s a very difficult thing to do,” Suga said. “I spent 10 years on this. I had many failures, but then I had two successes but they weren’t really useful, so that means failure for me. And then finally I came up with this flexizyme prototype, and I thought ‘this is it.’” Enter Kiichi Kubota, the business brain and co-founder who runs PeptiDream today. Together, they nailed down patents on the technology and worked on ways to make the process of discovering “hits” – the starting point for developing drugs – more efficient. By reducing the number of steps, the company cut the average time needed to discover them from about three days to four hours, according to Patrick Reid, PeptiDream’s chief science officer. That also lowered the potential for human error, he said. The Peptide Discovery Platform System has had strong interest from big pharma. Already, 16 of the most established names in the industry have signed agreements to work with PeptiDream to find hits for various diseases. The system can help discover drugs for pretty much anything, from cancer to neurological disease. Three firms, Bristol-Myers Squibb Co., Eli Lilly & Co. and Novartis AG, have gone a step further by licensing the technology to use in-house. PeptiDream’s revenue rose to 2.5 billion yen ($24 million) in the fiscal year ended June 2015. “These guys are different,” said Brian Heywood, chief executive officer of Taiyo Pacific Partners LP, which holds a 5 percent stake in PeptiDream despite being generally suspicious of biotech shares. Heywood says that PeptiDream doesn’t burn funds like some of its peers and is cash-flow positive. “This is one of those weird things where someone has something special that nobody can imitate. And it’s patented.” Not only that, PeptiDream kept part of its discovery for itself. Its system can create three types of drugs – peptide therapeutics, small-molecule medicines and what’s called peptide drug conjugates (PDCs). The first is mostly used for extracellular medicines, while the second, which are smaller, can permeate the cell. PeptiDream’s partnerships cover only those two. The third, PDCs as they’re called, are envisaged as a kind of smart drug. The peptide part will be used, for example, to home in on a cancer cell, which the conjoined drug will then attack. This contrasts with conventional treatments such as chemotherapy that kill other cells as well as the cancerous ones, resulting in hair loss, nausea and other symptoms. Reid and his team are focusing on this area within the company. “We carved that out,” Reid said. “The market is growing very rapidly. It’s one of the most rapidly growing areas of therapeutics.” Analysts, who are predominantly bullish on the stock, say one risk for PeptiDream is if big pharma starts to lose interest. They point to Pfizer Inc. canceling an agreement in 2013, and how shares tumbled on the news. “They have several partners, but we don’t know if the contracts will be extended indefinitely,” said Kiyokazu Yamazaki, an equity analyst at Ichiyoshi Research Institute Inc. who rates the shares a buy. “What people evaluate highly isn’t their creation of drugs in-house. It’s their contract revenue.” Shares surged 175 percent from November to a peak at the start of this month, capped by a 14 percent jump on June 3 after PeptiDream raised its annual profit forecast by 84 percent and said it got a second licensing payment from Novartis. The stock has tumbled more recently but even after the decline, it trades at 166 times earnings and 37 times book value. PeptiDream posted profit of 1 billion yen in the 12 months ended June 2015. The company moves to a new building near Tokyo Bay next year. Professor Suga remains an independent director and adviser, while his lab has moved on to other pursuits. His 8.6 percent stake is worth about $278 million, and he says he’s bought a house and filled it with guitars. Kubota, the president, now spends half his time talking to investors, and says he hopes Suga will win a Nobel Prize for his discovery one day. Meanwhile, across the corridor, Reid’s at work testing the boundaries of the new world of peptide drug conjugates. “We’ve developed a once-in-a-generation hit-finding platform,” Reid said. “It’s like sitting in a stack of gold every day.” Tom Redmond, Nao Sano, Bloomberg
Macau Daily Times라는 신문에서 PeptiDream의 스토리를 잘 소개한 것이 있어서 공유를 하는데 Suga Hiroaki (菅裕明) 교수는 본래 기타리스트를 꿈꿨지만 현실적인 이유로 신약개발을 하는 교수의 길을 가게 되었습니다. 미국에서 14년간 살다가 2003년에 동경대학 교수로 오게 되어서 Flexizyme을 발견한 후에 IP를 확보하고 동경대학에 있는 VC인 UTEC과 의논을 하면서 결국 PeptiDream을 창업하게 됩니다. PeptiDream은 일본 바이오텍의 몇개 안되는 유니콘 기업으로 $7 Billion Market Capitalization까지 올랐습니다.
Suga교수의 Flexizyme Project에 대해 사람들이 불가능하다고 얘기했지만 10여년의 기간 동안 수많은 실패를 딛고 결국 Flexizyme Prototype을 성공시키게 되었고 동경대학 교수가 되면서 Flexizyme을 완성하게 됩니다. 당시 동경대학 부교수였던 Patrick Reid교수가 CSO로 함께 참여를 했고 RaPID system을 3일 걸리던 것을 4시간만에 끝낼 수 있도록 발전시켰습니다. 16개의 글로벌 제약 바이오기업과 공동연구계약을 맺었고 BMS, Eli Lilly, Novartis는 연구계약을 넘어 기술을 라이센싱해서 자체적으로 할 수 있도록 시스템을 만들었습니다. 투자자들은 PeptiDream이 Cash-burn 없이 창업 2년차부터 수익을 얻는 비지니스 모델을 가진데 대해 특별한 투자기업이라고 얘기합니다. 공동창업자인 Kubota Kiichi(窪⽥規⼀)는 Suga Hiroaki (菅裕明)교수가 언젠가는 노벨상을 받을 것이라고 확신하고 있습니다. 저도 그렇게 생각합니다.
When he realized he wasn’t going to make it as a guitarist, Hiroaki Suga set out to find the origin of life, and ended up creating a new way to develop medicines. Many years spent fiddling with the building blocks of the universe – combining molecules to form compounds – led to Suga developing an enzyme that opened the door to a faster method of discovering drugs. PeptiDream Inc., the company he co-founded, has inked deals with many of the world’s biggest pharma firms, and shares have surged more than nine fold since listing in 2013. “Everybody comes to PeptiDream,” Suga, 53, said in an interview from his office deep in the main campus of the University of Tokyo. An electric guitar hangs from his wall. “Everybody probably accepts now that the technology we developed is very, very smart, very efficient,” he said. “I might go back to looking for the origin of life after I retire.” PeptiDream is part of a handful of Japanese biotech ventures that have grown into billion-dollar companies, which also includes Sosei Group Corp., the drug maker that now accounts for about 14 percent of the Mothers Index of smaller shares, and Euglena Co., which is trying to make jet fuel from algae. Like Euglena, it originated within Japan’s equivalent of Harvard, where Suga is a professor. In fact, PeptiDream is still based there today. The path to becoming a USD3.2 billion company started when Suga’s lab developed an artificial ribozyme, which he named flexizyme for its “promiscuous” ability to help amino acids couple to form peptides. Libraries of peptides – proteins made from a small number of amino acids – had been used by health-care companies for years to facilitate drug discovery, but they hadn’t been effective because they were unstable. Armed with flexizyme, the self-described research heretic Suga turned conventional wisdom on its head by making new libraries of a different type of peptides, often shaped more like a hula hoop than the spaghetti-type ones employed in the past. The steadier structure made them better at blocking the interactions between proteins that cause many diseases, according to Suga.
UTEC에서 PeptiDream의 창업부터 IPO까지 함께 한 VC인 Katadae Maiko(片田江 舞子) 박사가 PeptiDream President & CEO인 Kubota Kiichi(窪⽥規⼀)를 인터뷰한 것이 있습니다. 동경대학 (東京⼤学) Suga Hiroaki (菅裕明) 교수가 Katadae Maiko(片田江 舞子) 박사와 얘기하면서 회사 창업 가능성을 생각하게 되었고 Katadae Maiko(片田江 舞子) 박사가 Kubota Kiichi(窪⽥規⼀)를 CEO로 천거했다고 합니다. Kubota는 IP Strategy를 잘 설계해서 Flexizyme에 대한 특허와 FIT system에 대한 특허를 획득한 후 향후 2년간 RaPID 특허기술을 완성하는데 전력하게 됩니다. 이렇게 특허를 확보한 후 PeptiDream이 세계적으로 다른 회사들이 갖지 못한 독보적인 기술 플랫폼을 확보했다는 확신했다고 합니다. Suga 교수의 논문을 읽은 제약회사들이 PeptiDream에 공동개발을 청해왔을 뿐 특별히 PeptiDream이 제약회사들을 접촉하려고 한 적이 없다는 점이 좀 특이하고 의아하게 생각했습니다. 기술의 수준과 독보적인 IP가 중요하다는 것을 다시 한번 깨달았습니다. 창업 당시 Business Plan에 의하면 창업 6-7년차부터 손익분기점을 맞겠다는 계획이었지만 실제로는 창업한지 2년차부터 손익분기점을 넘어섰고 빠르게 성장해서 놀랐다는 얘기를 서로 하며 웃었군요. VC인 Katadae가 창업 초기부터 다른 VC들 처럼 하지 않고 파트너로 함께 했다고 Kubota 사장은 얘기했습니다.
In the natural world, there are creatures which are capable of producing substances unique to those species. In many instances in human history, such naturally occurring substances have been used as “miracle medicines” to save human lives. One class of such substances are “special peptides” produced by synthesizing “special amino acids” that do not occur naturally in the human body. PeptiDream was the first in the world to succeed in artificially synthesizing special peptides and linking it to drug discovery. PeptiDream has received a great deal of attention from the scientific and pharmaceutical industries and now, we are continuing our quest for medicines to cure incurable diseases around the world. The company was listed on the TSE Mothers in June 2013. How was PeptiDream able to commercialize a completely new drug discovery platform? We talk with the founder, President and CEO, Mr. Kiichi Kubota, and his companion from the founding period, UTEC partner, Dr. Maiko Katadae.
Kubota: There are 20 types of amino acids we possess in our body that have the l configuration. All proteins and peptides we synthesize in our bodies are made from these l-amino acids, its like we are playing LEGO.
On the other hand, in nature there are fungi, mosses, animals and plants which can produce special peptides from their own individual amino-acids. Often times the ‘miracle medicines’ that are discovered from rare species from the amazon or Tibet are these special peptides. The inception of our technology was an idea to make these special peptides available in test tubes rather than take a trip to the depths of the amazon forest or the Tibetan mountains.
Let’s review our science text books. Our bodies are made from several trillion cells. Each cell has 20 types of amino acids which it uses to synthesize its proteins and peptides. This synthesis occurs in the ribosome “the protein synthesis factory” from information obtained by copying the building instruction from the cell’s nucleus. If we can control the functioning of this ribosome, we can make proteins and peptides from selected amino acids and construct a practical drug discovery platform.
Kubota :The process through which the cell synthesizes its proteins or peptides have been long thought to be the “god’s work” or something that humans could not intervene with. However, if we use the “flexizyme technology” which our company’s co-founder Professor Suga invented, we can use the functioning of the ribosome to combine amino acids selectively. Even if the amino acid is specialized, we can handle the amino acids just like we would a normal one.
The flexizyme technology is a technology which “fools” the ribosome into combining amino acids and making proteins as we would intend it to. In addition, Peptidream has been able to not only synthesize specialized peptides but also to unlock the infinite opportunity of peptide drug discovery.
Kubota: Even if we are able to synthesize specialized peptides, the technology wouldn’t be so great if we could only create one or two. For drug discovery, we would need to be able to make various peptides at large volumes (a peptide library) and to screen for those peptides that would be appropriate to act as drugs. We were able to develop the ‘FIT system” which enables such library of specialized peptides, to the point where one test tube would contain a trillion peptides. Furthermore, we developed the ‘RAPID Display’ technology which allows for a fast and accurate screening of these billions and trillions of peptides. Combining these two technologies we established a drug discovery platform system ‘PDPS”. We now have alliances with global pharmaceutical companies and are at the forefront of drug discovery.
How will this special peptide platform make better our everyday lives? What will be made possible?Kubota: All diseases without cure are the addressable markets for the drugs discovered using our platform. Example of these include diseases which consistently rank high on the causes of deaths in developed countries such as cancer, diabetes mellitus, high blood pressure, other lifestyle diseases and their complications. Our dream is to stand up against these diseases through drug discovery. Fortunately, global pharmaceutical companies are also looking to cure diseases which have no cure. We are lucky that their needs and our intentions are aligned.
UTEC’s Dr. Maiko Katadae has seen Peptidream from its birth to the drug discovery technology of dreams that it is today. Peptidream sprouted from the day Dr. Katadae met Professor Suga’s flexizyme technology. Through the advice from the Technology Licensing Office of Tokyo University that this technology may be suited for development by a venture, Dr. Katadae decided to support the incorporation of Peptidream and the commercialization of its technology.
Katadae: When I first saw Professor Suga’s invention I was astonished to say the least. At that point, there was no candidate drug being produced by the flexizyme technology yet but I had firm belief that this technology would enable drug discovery that had never been seen before and decided to pursue the incorporation of the company with professor Suga and the Tokyo University TLO. I met Mr.Kubota in the autumn of 2005 and felt his personality and his business philosophy was a great fit to the company and introduced him to Professor Suga as a CEO candidate for the new company.
And so Peptidream was incorporated in 2006 with Mr. Kubota as CEO. Peptidream’s strength of course lied in its flexizyme technology. Mr.Kubota came up with an impressive IP strategy fully utilizing the uniqueness of the flexizyme technology. Peptidream hence became a unique existence that no other company could imitate. Currently, Peptidream possesses library building technology, FIT system, RAPID display screening system but these were all created through Mr.Kubota’s IP strategy.
Kubota: At first our IP was only the flexizyme technology. After that, we were granted the patent for the FIT system. However for the screening technology to screen which peptides would be useful as a drug I realized we had to in-license external IP. If a company tries to make up for a technology deficit through the in-licensing of another companies technology, the value of the existing technology IP falls to below half its original value. It was a hard pill to swallow but we decided to focus on the development of our own display technology. It took two years to produce our unique RAPID display technology. Peptidream was now able to be established as a drug discovery platform through the combination of the three patented technologies. We made sure that no one else could do the same drug discovery without the combination of these three uniquely patented technologies. We created a situation that if anyone wanted to perform special peptide discovery, they would have to sign a contract with us. Till this day, there are no other company anywhere in the world which has a platform that can perform from drug discovery to drug screening.
Peptidream currently has contracts with domestic partners Daiichi Sankyo, Teijin Pharma and international pharmaceutical companies as an alliance partners. A venture that came from a “zero start” and growing through partnership with large corporations, is the ideal form of a biotech venture. We think this was only possible due to IP strategy and in-house technological development.
Kubota: The reason we were able to pursue joint development schemes and form alliances with domestic and international pharmaceutical companies from the very early days of our companies is because we had our unique technological development of course, but also because we solidified our patent strategy and established a “winning pattern” for ourselves.
We incorporated the company in 2006. Back then, the center of the pharmaceutical industry was antibody drugs. However, many international pharmaceutical companies had been looking for candidates for the next drug development as they foresaw high competition. With this background, pharmaceutical companies who saw our publication on specialized peptide drug discovery started reaching out to us. We only responded to these offers. We did not proactively approach the pharmaceutical companies. When signing contracts with them, we would submit our standard fee table for each joint development, our strategy was to always maintain a bullish attitude.
These strategies eventually turned to fruit and peptidream was able to be a profitable business from its second year onwards. We currently have contracts as alliance partners with 13 global pharmaceutical companies.
Kubota: The first impression I had of Dr. Katadae was that she was a technology enthusiast rather than someone at a venture capital firm.
Dr. Maiko Katadae, now a partner at UTEC had been with Peptidream since its very inception. The first task Dr. Katadae and Mr. Kubota did together was not to invest but to make an office at the University of Tokyo, cleaning it and bringing chairs and basic office supplies together. The investment into Peptidream happened in 2008.
Katadae: When I first saw the felxizyme technology I thought that “this technology will be a success or rather I want to make it a success!”. That’s why we invested in Peptidream but do you remember the business plan we first made together? We were planning about $10M USD sales in the company’s 6th or 7th year.
Kubota: hmm, the documents are still there but I don’t remember the numbers (laughs).
Katadae: We were at a stage where we did not even know if the business model would work. “We probably said one project should bring about this much revenue, and thought how many of those we needed to achieve our goal”. When we looked back, those numbers were so far off. Peptidream’s business model is one which presumes that pharmaceutical companies would be willing to reveal information about their drug targets. In simpler terms, it is assumed that pharmaceutical companies would reveal information usually kept very secret. We received a lot of comments in the line of “there is no way that is possible”. I remember being very frustrated at this but now I think of it as “We were able to prove a business model which people thought impossible” which I find very meaningful.
Kubota: Dr. Katadae has always been with us, from when we were setting up our first office until our IPO. The perspective to see a venture capitalist as not someone who would just offer money but as someone who chases dreams with you is a very important perspective in making a successful business.
Katadae: At the start, venture capitalists and entrepreneurs sit at opposite sides of the table. However, in the process of investing, I place an emphasis on how close we can sit with the entrepreneurs on the same side of the table. Venture capitalists until they invest are simply looking to buy shares at a cheap rate and then sell at a high price, hence our incentives are the opposite of an entrepreneurs’. The important thig is that trust and closeness are built in the process of having many discussions with the entrepreneurs that when we realize, we have ended up on the same side.
Kubota: Well, in our case you were literally sitting next to me from the very start.
Peptidream went public on the Tokyo Mothers Exchange in June of 2013. Peptidream’s share was valued at 7900 yen, 2500 yen over the public price. It’s market capitalization has surpassed ~$6 billion USD. Mr. Kubota dreams of having the first drug go commercial with the initials “PD”
Kubota: I do have a wish to make a drug that has the “PD” initials. However, at the base of this wish is my motivation to work that alleviates the suffering of patients.
Provide novel drugs for patient without cures, that is always our mission.
Suga Hiroaki 교수는 Flexizyme이라는 새로운 효소를 개발하였습니다. 이에 대한 리뷰는 2011년에 Accounts of Chemical Research에 실린 바 있습니다. Flexizyme은 In Vitro Selection으로 Artificial Amino Acids를 tRNA에 결합시키는 Artificial Ribozyme을 의미합니다.
FIT (Flexible In Vitro Translation) System은 Flexizyme이라는 Novel Ribozyme을 이용해서 tRNA에 Unnatural amino acids를 결합시킨 후 In-Vitro Translation을 통해서 “Reprogrammed Translation을 통해서 “Thioether-closed Macrocyclic Peptides (tcMPs)를 만들고 이것을 mRNA Display로 만드는 것입니다.
RaPID (Random nonstandard Peptides Integrated Discovery)는 결국 FIT와 mRNA Display를 결합시킨 시스템을 말하는 것입니다.
최근 5년간의 공동연구계약 상황을 보면 Macrocyclic Peptides와 Peptide-Drug Conjugate (PDC) 분야로 나뉘어져서 비교적 골고루 공동연구계약이 이루어지고 있는 것을 볼 수 있습니다. Genentech의 경우에는 Macrocyclic Peptides와 PDC 분야 모두에 공동계약을 하고 있습니다.
2023년 11월 현재 Peptidream의 Pipeline은 아래와 같습니다. 아직까지는 임상1상이 가장 앞선 단계인 Early-stage입니다. Hit Identification까지는 비교적 빠르게 진행할 수 있지만 역시 Drug Candidates를 얻는데까지 Lead Optimization은 시간이 상당히 걸리는 것 같습니다. 창업한지 18년차가 되었습니다. 지속적인 성장을 통해 언젠가 PeptiDream도 자체적인 개발 프로그램을 갖게 되기를 기대합니다.
2025년 11월 17일 (월요일)
펩티드림에 대해서 글을 쓴 지 1년여가 지났군요. 작년에 글을 쓸 때에는 긍정적인 부분을 많이 쓰고자 노력을 했는데 이번에는 수가 히로아키 교수가 펩티드림에 대한 소회를 인터뷰한 것이 있어서 좀 남기려고 합니다.
University and government venture funds play a much larger role in Japan than they do in Western countries. Yet we see fewer biotechnology startups here compared with, say, the United States, which is home to eight of the top 10 highest-funded ventures. Why?
“If you have $10 million, you will just burn through it,” Suga said, adding that less capital will keep you focused and get results that can lead to bigger things...With limited funds, “You need to really develop technology that will allow you to collaborate with big pharmaceutical companies,” Suga explained.
This unusual approach has worked well for PeptiDream, so why don’t we see more biotech startups succeeding this way in Japan?
Suga said there are several reasons.
“The first is that venture capitalists are not investing in risky companies, and biopharmaceutical companies are high risk,” he explained. “If you are developing business software, after six months, you know if it isn’t working.
“The second reason is that Japanese society prefers to go with what’s known,” he continued. In this case, it means that talent heads for the largest pharmaceutical companies, which are seen as stronger and a safe harbor. “For example, all my students go to big pharma. They don’t go to PeptiDream.”…Large Japanese companies tend to have little interest in helping smaller ones.
The third obstacle that Suga cited is the fact that many startups in Japan are research units that have been spun off from large companies that chose to leave Japan. “They had a very good team here, so they decided to spin off. They already have a background from big pharma and continue doing [what they were doing],” he explained. “That means that they aren’t hugely different from the big companies.”
Suga 교수님은 일본의 문화적 습성에 때문에 대학으로 부터 스타트업이 생길 수 없다고 말씀하십니다. 즉,
VC가 biotech에 투자하는 것을 꺼리고
일본 사회가 큰 회사를 선호하는 경향이 있으며
일본의 스타트업 대부분은 일본 대기업에서 분사한 회사로서 일본을 떠나려는 회사들이기 때문이다.
Novo Nordisk는 덴마크에서 Insulin을 생산하는 회사로 1923년에 창업해서 이제 100년이 넘는 역사를 가진 제약회사입니다. 2023년 현재 전세계 Top15 거대제약회사 중의 하나이고 당뇨병 치료제로만 이런 성적을 거두고 있습니다. Novo Nordisk가 최근에 주력하는 약물은 Semaglutide라는 Glucagon-Like Peptide 1 (GLP-1) Receptor에 작용하는 펩타이드 약물로서 주사제인 Wegovy, Ozempic이 있고 경구용 약물인 Rybelsus 이렇게 세가지 품목을 판매하고 있습니다.
그리고 현재 Weight loss 약으로 50mg oral Semaglutide의 OASIS 임상3상이 진행 중입니다.
Glucagon-Like Peptide-1 (GLP-1)은 31개 아미노산으로 연결된 펩타이드인데 이것의 발견이 있기 전에 Mount Sinai 병원의 John Eng 박사가 한 1990년대 초반의 연구부터 시작을 합니다. John Eng 박사는 남아메리카에 서식하는 도마뱀인 Gila monster의 타액에서 Exetadin-4라는 펩타이드를 발견하게 되는데 이 펩타이드가 혈당을 낮추는 효과가 있다는 것을 발견하게 되고 이 펩타이드와 유사한 인간 펩타이드 GLP-1을 발견하여 세상에 이 사실을 알리게 됩니다.
아래의 논문은 John Eng박사가 1997년에 GLP-1에 대한 연구결과를 Journal of Biological Chemistry에 발표한 논문입니다.
John Eng박사는 이 연구결과를 신약개발에 활용할 수 있다는 확신을 가지고 정부기관, 제약회사 등을 찾아다니며 설득을 했지만 결국 큰 소득을 얻지 못하게 되고 작은 바이오텍인 Amylin Pharmaceuticals에 이 기술을 팔았는데 2005년에 Eli Lilly-Amylin Pharmaceuticals가 BYETTA(TM) (Exenatide) Injection의 FDA 승인을 얻습니다.
Exenatide는 39개의 아미노산으로 구성된 펩타이드인데 주사제로 개발이 되었고요 새로운 당뇨병 치료제의 개발이 필요했습니다. 또한 GLP-1이 당뇨 뿐만 아니라 체중감소에도 효과가 있는 기전인 것이 밝혀지며 Novo Nordisk는 새로운 GLP-1 R Agonist 개발에 뛰어들게 되고 Semaglutide라는 약물을 얻게 됩니다.
John Eng박사의 연구와 Semaglutide의 개발 승인까지의 이야기는 Hilary Brueck이 Business Insider India에서 잘 다뤄주고 있습니다.
What does the Gila monster have that we don’t have? The key to more effortless weight-loss, apparently.
It turns out the venom of a small, Southwestern lizard — the only venomous lizard in America — played a critical role in developing a whole new class of blockbuster anti-obesity drugs, called GLP-1s.
One of the newest GLP-1s is called semaglutide. It’s sold under the brand names Ozempic and Wegovy — and it is taking Hollywood by storm. Rising demand for these types of drugs, which mimic key hormones that tell us to feel full, have led to severe shortages of GLP-1s in recent months.
But before semaglutide became the darling shot of Hollywood, scientists discovered that compounds in the venom of Gila monsters could help drug developers make better diabetes medications than they’d ever had before.
Gila monster hormones can regulate blood sugar very well
It all started back in the early 1990s, when government researcher Dr. John Eng discovered that Gila monsters have a special hormone in their venom. The hormone is quite similar to a hunger-regulating hormone humans harbor in the small intestine, which helps control blood sugar levels.
In people, it’s called glucagon-like peptide-1. In Gila monsters, Eng named it exendin-4.
Exendin-4 degrades more slowly than the human form of GLP-1, lasting for hours instead of minutes. That means it’s a much better model for drug development, since it wouldn’t be practical to take a drug dozens of times a day.
At first, Eng tried to point this remarkable feature of Gila monster spit out to pharmaceutical makers and the government. He shopped his idea around at the Department of Veterans Affairs, where he worked at the time, as well as several different pharmaceutical companies, but didn’t have much success. In the end, he patented the molecule in 1995, and licensed the discovery to a now-defunct biotech startup called Amylin.
Amylin used Eng’s Gila monster research to create a synthetic hormone, called extenatide. Extenatide was approved by the Food and Drug Administration (FDA) in 2005 to treat type 2 diabetes. It’s still used by hundreds of thousands of children and adults with diabetes today.
A safe obesity treatment that slows digestion and curbs cravings
Extenatide was the very first GLP-1-mimicking drug. It ushered in a whole new class of diabetes medications that are arguably safer, and more effective, than previous treatments were. More recently, GLP-1s have been designed to target obesity, too.
Today’s GLP-1s work to help people lose weight because they mimic a hormone our small intestine makes naturally, which regulates hunger in several different key ways.
When a patient’s blood sugar levels are high, GLP-1 drugs send signals to their pancreas to secrete more insulin — but the hormone-mimicking doesn’t stop there. GLP-1s also send signals to a person’s brain, telling their body to feel fuller with less food. Finally, GLP-1s slow down digestion, changing the way a person’s body turns food into energy.
Originally, patients had to take extenatide twice a day. But, over time, newer, more advanced GLP-1s have come to market, with even longer release times (no offense, Gila monsters).
We need animals to create medical breakthroughs like Ozempic, scientist says
Today, most GLP-1s are injected once a day, or just once a week. But they arguably wouldn’t be here if it wasn’t for Eng’s work — which created the very first GLP-1 drug.
In a statement to Insider, Novo Nordisk, the company that makes Wegovy and Ozempic, said that Gila monsters and the discovery of exendin-4 “did not have anything to do with our decision to develop long-acting GLP-1 receptor agonists” for obesity, because “that was based on GLP-1 biology in humans.”
Semaglutide의 개발에 대한 결과는 2015년 Journal of Medicinal Chemistry에 보고를 했습니다. 논문 링크는 아래에 보냅니다. Liraglutide라는 Lead compound로 부터 지속성을 늘리기 위한 다양한 Lead Optimization을 통해 결국 Semaglutide라는 약물을 얻게 되었습니다.
Liraglutide와 Semaglutide의 구조는 아래에서 보이듯이 상당한 Modification이 되었습니다. 세번째 아미노산인 Alanine이 DDP-4에 쉽게 분해되는 것을 막기 위해 Dimethylglycine으로 치환하고 다양한 Lipophilic linker를 집어넣어서 Stability를 크게 늘이는 노력을 기울인 것을 알 수 있습니다.
Liraglutide와 Semaglutide의 개발에 대한 스토리는 Novo Nordisk 연구진이 2019년 Frontiers in Endocrinology논문에 비교적 상세히 실었습니다.
경구용 Semaglutide는 SNAC (Sodium N-(8-[2-Hydroxylbenzoyl] Amino) Caprylate)라는 Absorption Enhancer를 함께 넣어준 새로운 Formulation Drug입니다. SNAC formulation은 본래 Emisphere Technologies가 보유한 기술로서 2007년부터 Novo Nordisk와 공동으로 Oral Semaglutide (Rybelsus) 개발에 사용을 하던 것을 소유권을 얻기 위해 Emisphere를 총 $1.8 Billion에 인수하게 됩니다.
Novo Nordisk A/S today announced that the company has entered into a definitive agreement to acquire Emisphere Technologies Inc. (Emisphere), a drug delivery company with proprietary technologies, such as the Eligen® SNAC technology, that enable oral formulations of therapeutics.
Novo Nordisk and Emisphere have collaborated since 2007 and Emisphere’s proprietary drug delivery technology Eligen® SNAC is used by Novo Nordisk under an existing licence agreement in the oral formulation of Novo Nordisk’s GLP-1 receptor agonist semaglutide, which is marketed and sold under the brand name Rybelsus®.
Under the terms of the agreement, Novo Nordisk will acquire all outstanding shares of Emisphere for USD 1.350 billion. As part of the transaction, Novo Nordisk will also acquire related Eligen® SNAC royalty stream obligations owed to MHR Fund Management LLC (MHR), the largest shareholder of Emisphere, for USD 450 million. Consequently, the total acquisition price is USD 1.8 billion.
With these acquisitions, Novo Nordisk eliminates its future royalty obligations to Emisphere and MHR and obtains full access to the Eligen® SNAC technology platform thereby enabling Novo Nordisk to expand the portfolio of oral biologic pipeline assets across therapy areas.
The transaction will be debt financed and will not impact Novo Nordisk’s previously communicated operating profit outlook for 2020 or the ongoing share buyback programme. The acquisition is expected to have a net negative impact on operating profit of less than one percent in 2021 and broadly neutral to positive impact in the following years.
The acquisition of Emisphere provides Novo Nordisk full ownership of the Eligen® SNAC technology, which has been successfully used under a licence agreement to develop the first oral biologic, Rybelsus®” said Mads Krogsgaard Thomsen, executive vice president and chief scientific officer of Novo Nordisk. “We intend to apply and further develop the technology and use it on current and future pipeline assets with the aim of making more biologic medicines orally available for patients”.
The transaction is subject to customary closing conditions, including approval by Emisphere shareholders and the expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976. MHR and certain other shareholders of Emisphere, collectively owning a majority of the Emisphere shares, have agreed to vote their shares in favour of the transaction.
Novo Nordisk is represented by Davis Polk & Wardwell LLP as legal advisor and Evercore as financial advisor.
About Eligen® SNAC Carrier Technology Eligen® SNAC technology enables drug therapies to be provided in a tablet formulation with an absorption-enhancing excipient. Emisphere created Eligen® SNAC technology, its proprietary oral drug delivery platform, to facilitate the absorption of small and large molecules without altering their chemical form, biological integrity or pharmacological properties. Notably, the technology enables the transport of therapeutic molecules including large peptides and proteins across biological membranes such as those of the gastrointestinal tract.
About Emisphere Emisphere is a drug delivery company that utilises its proprietary technologies to develop new oral formulations of therapeutic agents. For more information, please visit Emisphere’s website at www.emisphere.com.
SNAC oral delivery fomulation 에 대한 좋은 리뷰가 있어서 두개를 올립니다. 첫번째는 영국 University of Wales 논문입니다.
Novo Nordisk today announced that the U.S. Food and Drug Administration (FDA) has approved Rybelsus® (semaglutide) tablets 7 mg or 14 mg for adults with type 2 diabetes that along with diet and exercise may improve blood sugar (glucose).1 Rybelsus® is the first and only glucagon-like peptide-1 (GLP-1) analog in a pill and a new option for adults with type 2 diabetes who are not achieving their A1C goal with current antidiabetic treatment.
Type 2 diabetes is a global public health issue that impacts more than 28 million people in the U.S. alone.2 Despite existing treatment options, many adults with type 2 diabetes have poorly managed blood sugar that can increase the risk of developing serious diabetes-related complications.2-3
“GLP-1 receptor agonists are effective medications for people with type 2 diabetes but have been underutilized in part because they have, until now, only been available as an injectable treatment,” said Vanita R. Aroda, MD, Director of Diabetes Clinical Research, Brigham and Women’s Hospital, Boston, MA and a PIONEER clinical trial investigator. “The availability of an oral GLP-1 receptor agonist represents a significant development and primary care providers, specialists and patients alike may now be more receptive to the use of a GLP-1 therapy to help them achieve their blood sugar goals.”
The approval of Rybelsus® is based on results from 10 PIONEER clinical trials, which enrolled 9,543 participants and included head-to-head studies of Rybelsus® vs. sitagliptin, empagliflozin and liraglutide 1.8 mg.4 In the trials, Rybelsus® reduced A1C and, as a secondary endpoint, showed reductions in body weight. The most common adverse reactions in the PIONEER trials, reported in ≥5% of patients, were nausea, abdominal pain, diarrhea, decreased appetite, vomiting and constipation. The types and frequency of the adverse reactions were similar across trials.1,5-7
“People living with type 2 diabetes deserve more innovation, research and support to help them achieve their individual A1C goals,” said Todd Hobbs, vice president and U.S. chief medical officer of Novo Nordisk. “With Rybelsus®, we have the opportunity to expand use of effective GLP-1 receptor agonist therapy by providing adults with type 2 diabetes an oral medication which was previously only available as an injection to help with managing their blood sugar.”
Rybelsus® is approved for once-daily use in two therapeutic doses, 7 mg and 14 mg, and will be available in the U.S. beginning in Q4 2019. Initial supply of Rybelsus® will come from manufacturing facilities in Denmark; however, future supply for Rybelsus® will come from manufacturing facilities in the U.S. In 2015, Novo Nordisk made a strategic investment to build a new manufacturing facility in Clayton, NC to prepare for the future demand for Rybelsus®. Additionally, earlier this year Novo Nordisk acquired a tableting and packaging facility in Durham, NC to meet anticipated supply needs for Rybelsus®.
Novo Nordisk is working with health insurance providers with a goal of ensuring broad insurance coverage and patient access to the product. A savings card program will be available at the time of launch for eligible commercially-insured patients to keep out of pocket costs down to as little as $10 a month.
The U.S. FDA is still reviewing Novo Nordisk’s new drug application (NDA) for Rybelsus® seeking an additional indication to reduce the risk of major adverse cardiovascular events (MACE) such as heart attack, stroke, or cardiovascular death in adults with type 2 diabetes and established cardiovascular disease (CVD). A decision is expected in Q1 2020.
Rybelsus® is currently under review by several regulatory agencies around the world, including the European Medicines Agency and the Japanese Pharmaceuticals and Medical Devices Agency.
What is Rybelsus®?
Rybelsus® (semaglutide) tablets 7 mg or 14 mg is a prescription medicine for adults with type 2 diabetes that along with diet and exercise may improve blood sugar (glucose).
Rybelsus® is not recommended as the first choice of medicine for treating diabetes
It is not known if Rybelsus® can be used in people who have had pancreatitis
Rybelsus® is not for use in people with type 1 diabetes and people with diabetic ketoacidosis
It is not known if Rybelsus® is safe and effective for use in children under 18 years of age
그리고 Rybelsus는 first-line type 2 diabetes option으로 FDA label update가 되어 이제 많은 당뇨환자들에게 쓰일 수 있게 되었습니다.
The U.S. Food and Drug Administration (FDA) has approved a label update for Rybelsus® (semaglutide) tablets 7 mg or 14 mg, allowing use as a first-line treatment option for adults with type 2 diabetes who have not previously taken a diabetes treatment. This update removes a previous limitation of use that stated the medication should not be used as the initial therapy for treating patients with type 2 diabetes. Initially approved by the FDA in 2019, Rybelsus® is the first and only GLP-1 analog in pill form and is indicated, along with diet and exercise, to improve glycemic control for adults with type 2 diabetes.1,2
“The removal of the limitation of use is an important step forward for people living with type 2 diabetes and provides the option for Rybelsus® to be taken earlier,” said Dr. Aaron King, Family Medicine and Diabetes Specialist. “By taking Rybelsus® first, people with type 2 diabetes, in conjunction with their care teams, are now able to utilize this medicine early in their diabetes treatment journeys.”
Rybelsus® works differently than other diabetes pills to lower blood sugar in three ways: by increasing the release of insulin from the pancreas when blood sugar is high, decreasing the release of sugar from the liver, and slowing the process of food leaving the stomach after eating.1,2 Rybelsus® comprises a unique co-formulation of semaglutide and an absorption enhancer called SNAC (sodium N-(8-[2-hydroxybenzoyl] amino) caprylate), which facilitates absorption of semaglutide in the stomach, making it possible to provide semaglutide as a pill.4
“In the U.S., hundreds of thousands of people with type 2 diabetes have been prescribed this medicine as part of their type 2 diabetes treatment regimen to help lower their A1C,” said Doug Langa, executive vice president, North America operations and president of Novo Nordisk. “As Novo Nordisk marks 100 years of commitment and innovation in diabetes care, Rybelsus® remains a pivotal part of our portfolio, making history as the first oral GLP-1 receptor agonist and helping to fuel our mission to improve the lives and health of people living with diabetes.”
Novo Nordisk works with health insurance providers to ensure broad insurance coverage and patient access to Rybelsus®. Eligible, commercially insured patients may pay as little as $10 for a one- to three-month prescription of this medicine.
For more information about Rybelsus®, visit Rybelsus.com. For health care professionals, please visit RybelsusPro.com.
What is RYBELSUS®?
RYBELSUS® (semaglutide) tablets 7 mg or 14 mg is a prescription medicine used along with diet and exercise to improve blood sugar (glucose) in adults with type 2 diabetes.
It is not known if RYBELSUS® can be used in people who have had pancreatitis
RYBELSUS® is not for use in people with type 1 diabetes
It is not known if RYBELSUS® is safe and effective for use in children under 18 years of age
2023년 6월에 Novo Nordisk가 ADA (American Diabetes Association)에서 발표한 자료가 아래에 링크했습니다. Semaglutide 이외의 Novo Nordisk 약물에 대한 내용도 함께 있습니다.
현재 Oral Semaglutide SNAC formulation 50mg에 대한 OASIS 임상3상이 진행 중인데 이에 대한 결과는 The Lancet 2003년에 발표했고 아마도 곧 승인되어 Wegovy의 weight loss 주사제로 부터 새로운 경구용 체중감소제의 시장이 곧 열릴 것 같습니다.
보통 은퇴전문가들이 은퇴에 필요한 자산을 얘기할 때 “20억원이 필요하다”, “30억원이 필요하다” 이런 말들을 해서 은퇴를 앞둔 사람들을 불안에 떨게하는 공포마케팅을 하는데 그 정도 돈이 있으면 이미 자산가입니다. KB금융지주 한국부자 보고서에 의하면 10억 이상 금융자산을 가진 사람이 “자산가”로서 부자입니다.
전체 자산이 30억원 미만인 사람을 보면 33% 정도 그러니까 10억원이 금융자산이고 30억 이상이 되어도 금융자산이 50%가 안되니 결국 30억원 금융자산이 있으려면 자산이 근 70억원은 있어야한다는 실현이 거의 불가능한 비현실적인 답에 접근하게 됩니다.
그럼 투자에 대한 부자들의 실력은 어떨까?
먼저 금융상품 투자를 보면 만기환급형 보험이 가장 수익이 난 것을 알수 있고 그 다음이 주식 > 펀드 > 채권 순이었습니다. 그러니까 부자라 하더라도 금융투자실력은 그리 높지 않은 것으로 나타났습니다.
부동산 투자실력은 어느 정도일까요?
투자용 부동산보다는 거주용 부동산에서 수익이 좀더 나았고 투자형 부동산도 주로 아파트 투자가 그나마 가장 안전한 투자인 것으로 보입니다.
그러니까 부자라 하더라도 투자할 자금이 넉넉한 것이 아니고 투자실력도 아주 훌륭한 것이 아니니 모두 일을 해야한다는 것이 결론입니다.
17일 KB금융지주 경영연구소가 발간한 ‘2023 한국 부자 보고서’에 따르면 이 부자들은 총 2747조원의 금융자산과 2543조원의 부동산자산을 보유했다.
자산 규모별로는 부자의 91.2%(41만6000명)가 10억~100억원 미만의 금융자산을 보유한 ‘자산가’로 분류됐다. 보유 금융자산이 100억~300억원 미만인 ‘고자산가’는 6.9%(3만2000명), 300억원 이상의 금융자산을 가진 ‘초고자산가’는 1.9%(9000명)를 차지했다. 초고자산가는 전체 우리나라 인구의 0.02%를 차지했다.
다음으로 보려는 보고서는 우리금융그룹이 내놓은 2023년 서울 부자 보고서입니다. 우리금융의 보고서는 서울에 사는 부자에 대해서 조사한 것이라 흥미롭습니다.
서울부자의 순자산은 60억 정도이고 이 중 40억이 부동산, 20억이 금융자산이군요. 여기에서도 현금을 만들 수 있는 금융자산은 20억 정도로 은퇴전문가들이 말하는 은퇴금융자산의 최저액을 서울 부자라 해도 도달하기 쉽지 않다는 것입니다.
서울 부자들은 거의 모두 돈을 벌고 있습니다. 사업소득이 근로소득보다 높은 것으로 나타나서 자신의 사업체를 운영하고 있다고 할 수 있겠죠. 직업구성 중에서 은퇴한 비율은 10% 이하이고 거의 80%가 자신의 사업을 하고 있군요.
서울부자들의 3년뒤 자산목표는 현재에 비해 아주 크지 않고 그 자산을 마련하는 수단으로는 사업소득이 가장 높았고 (68.3%), 투자의 경우는 사업소득 (42.2%) > 부동산투자 (41.4%) 순으로 대부분 자신의 사업을 통한 소득 창출을 하거나 부동산 투자로 자산을 조금씩 늘려가는 것으로 나타납니다. 그러니까 부자들은 큰 이익을 향해 투자한다기 보다는 상당히 안전한 투자 성향을 가진다는 것을 볼 수 있죠.
KB금융의 한국 부자 보고서는 전국을 대상으로 하는데 총자산이 대략 30억 정도에 10억 정도 금융자산을 가진 것으로 나타났고 우리금융의 서울 부자 보고서는 순자산이 대략 60억 정도에 20억 정도 금융자산을 가진 것으로 나타났습니다.
이런 금융자산은 소위 은퇴전문가들이 은퇴를 위해 필요하다고 하는 최소액에 해당합니다. 서울 부자들은 자신의 사업소득이나 부동산 투자소득으로 3년에 10% 정도 자산이 증식되는 목표를 가지고 사는 것으로 나타났습니다.
이 보고서들을 기준으로 보았을 때, 부자라고 해서 은퇴하고 놀면서 살고 있지 않고 투자를 해서 대박을 노리기 보다는 스스로 일을 해서 돈을 벌거나 부동산을 통한 일정한 자산 증식에 목표를 두는 것으로 나타났습니다. 바람직하죠.
일을 하지 않는 방법은 별로 없다는 것이 이 자료들을 읽은 후의 저의 생각입니다. 다들 열심히 일합시다!
Harrison Ford가 주연한 영화 “Extraordinary Measures”는 John F. Crowley의 이야기를 바탕으로 한 실화입니다.
“Extraordinary Measure“라는 영화는 John Crowley와 가족들의 삶을 담은 책 “The Cure: How a Father Raised $100 Million–and Bucked the Medical Establishment–in a Quest to Save His Children.“을 바탕으로 하고 있습니다. 이 책은 풀리처 상을 수상한 Greeta Anand가 썼습니다.
세자녀 중 두 아이가 태어난지 얼마 지나지 않아 Pompe Disease라는 근무력증 희귀질환을 앓고 있는 것을 알게된 아빠 John F. Crowley는 아이들의 치료를 위해 백방으로 수소문을 한 끝에 William Canfield 박사를 찾게 되고 그와 창업을 제안하며 Novazyme Pharmaceuticals를 설립합니다.
그의 삶에 대한 이야기는 Global Genes Blog에도 자세히 담겨 있습니다. 지금부터 John Crowley가 2살이 되기 전에 죽을 것이라는 두 자녀를 20대 성인이 될때까지 살 수 있도록 하기까지 어떠한 삶을 살았는지 나누고자 합니다.
John Crowley’s life is literally a Hollywood movie. His journey to save his children’s life was turned into the movie “Extraordinary Measures” starring Brendan Fraser and Harrison Ford, and his family has been featured in a number of magazine and newspaper articles, and in two books. John changed his career path to help find a cure for the disease that could end his children’s life, and to help others diagnosed with rare diseases, and along the way, has inspired many others with his leadership skills.
In March 1998, when John’s daughter Megan was 15 months old, she and her newborn brother, Patrick, were both diagnosed with Pompe disease. John and his wife Aileen were told their two younger children would not live past their second birthday.
Pompe disease is a neuromuscular disorder caused by a genetic mutation that prevents GAA (acid alpha-glucosidase) production. The result is a toxic buildup of glycogen which in turn causes a number of symptoms, including muscle weakness and swelling of vital organs, and ultimately, respiratory issues and heart failure. Symptoms can be managed with infusions of Lumizyme, which breaks down the glycogen, but it does not treat the cause of the buildup.
When John learned about the prognosis for his children, he felt he had to do more than just sit and watch their health decline. He was well educated, with a bachelor’s degree in foreign service from Georgetown University, a law degree from Notre Dame, and a Harvard MBA. His left his career in management consulting to take a position with a pharmaceutical company in 1998. In 2000, John co-found Novazyme Pharmaceuticals, a startup biotech, with glycobiologist, William Canfield, MD, PhD. Cash advances on credit cards and home equity line of credit on the Crowley’s home helped fund the start up. The gamble would be worth it in the long run.
Novazyme was bought by Genzyme within a couple of years. Genzyme has successfully received FDA approvals for treatments for rare diseases, and eventually created Myozyme, which was a life-saving medicine for Pompe disease. It was not an easy process for Megan and Patrick though. The Genzyme team thought they would have better success with infants to test the drug, and the Crowley children were already toddlers at this point. Even when Genzyme agreed to test the drug on older children, the Crowley’s children did not quality. Finally, Genzyme saw an opportunity to create a clinical trial just for Megan and Patrick, because while both children have an identical genetic mutation, Patrick is affected more than Megan. This “sibling test” gave Genzyme the opportunity to study why the treatment was more effective for some children than others. First, John had to resign from the company in order to avoid any conflicts for Megan and Patrick to be eligible for a clinical trial.
“We hadn’t seen her [Megan] smile in two years. After the first couple of months, we started to notice that she was smiling again.” John shared, “That was the first sign to me that there was some hope.”
Within the first few months of treatment, Megan and Patrick gained muscle strength, and their enlarged hearts returned to the normal size. As the study theorized, Megan did respond better to the treatment than Patrick, but both showed signs of improvement. That life-saving drug, Myozyme, was redeveloped into Lumizyme, and now, more than 3,000 people diagnosed with Pompe worldwide receive the treatments.
John went on to found Amicus Therapeutics, a biotech company that develops treatments for those diagnosed with rare diseases, including new treatments for those with Pompe disease. He has been a passionate advocate for providing universal access to treatments for those with rare diseases, as well as championing needs for those with disabilities.
“In our view, nobody in the world should ever be denied our medicine,” John said. “So we have to figure out creative ways to get it to everybody.”
John was introduced to Global Genes by founder Nicole Boice in 2009 when she was in the planning stages for the non-profit. John and some other contributors helped Nicole to envision the final product as an umbrella organization in the rare disease community. John became a founding board member in 2010 when Global Genes was founded. John told Nicole, “I love your passion, and I love the vision here. There’s an unbelievable opportunity to fill a huge void.”
As a researcher, John is still seeking new and better treatments through his work with Amicus, as a board member for Global Genes, and a former board member for the Make-A-Wish Foundation. As a father, John knows that his children are all adults now, and their medical decisions are their own to make. The Crowley’s oldest child, John Jr., was diagnosed with dyslexia, ADHD and Asperger’s syndrome as a child, so the Crowley family has had to navigate many diagnostic and treatment journeys throughout their kids’ childhoods.
“I think I did my job,” John said. “As a dad, I did what I had to do. I don’t think that makes me a hero.”
In addition to all of his work in the world of rare diseases and being a crusader for those living with disabilities, John has also become known as an authority in leadership, and has spoken to numerous groups about his experiences as a leader in challenging situations. Among his many speaking engagements, John spoke at the 2020 commencement at his alma mater, Notre Dame, at the Rothman Institute of Entrepreneurship at Fairleigh Dickinson University, and at the University of Georgia’s Terry College of Business for the Mason Public Leadership Seminar.
Pulitzer Prize-winning reporter, Geeta Anand, captured the Crowley’s odyssey from diagnosis to finding a treatment, first in a Washington Post article, then, in a book The Cure: How a Father Raised $100 Million–and Bucked the Medical Establishment–in a Quest to Save His Children.The book was the inspiration for the movie, Extraordinary Measures, starring Harrison Ford, Brendan Fraser and Keri Russell.
Imagine what the world would be like if John had not taken the extraordinary measures for his children to save their lives, and in turn affect the lives of thousands of others with Pompe disease, and all of the other lives of those diagnosed with rare diseases and living with disabilities that he has affected.
John Crowley는 Novazyme을 설립할 때 자신의 집을 담보로 자금을 마련하여 Preclinical 연구펀딩을 합니다. 그리고 그 데이타를 바탕으로 $8 Million의 Series A를 함으로써 임상에 진입합니다.
Novazyme Pharmaceuticals has completed an $8 million private placement of Series A preferred stock, the Oklahoma City company said Tuesday.
The placement was co-led by: Catalyst Health & Technology Partners (Boston); HealthCare Ventures (Princeton); and Perseus-Soros Biopharmaceutical Fund (New York). Novazyme will use the funds to accelerate the clinical development of its lead lysosomal storage disease programs.
Novazyme has developed a series of proprietary technologies that have been shown in pre-clinical studies to greatly enhance uptake of replacement enzymes for lysosomal storage diseases. Virtually all of these diseases share a common biologic pathway for uptake through mannose-6 phosphate receptors. Novazyme’s core technology targets enzymes to these receptors. It is applicable to enzyme therapies for all of the lysosomal diseases that share the mannose-6 phosphate pathway. Enhanced enzyme uptake is widely viewed as the key to the next generation of drugs to treat these rare diseases. With more efficient uptake of replacement enzymes, patients may benefit by greater response to these therapies and by reduced side effects, such as antibody responses.
“Novazyme represents a unique opportunity to invest in a company that combines a powerful core platform technology with its own internal drug development programs,” said Dennis J. Purcell, managing director at Perseus-Soros. “The untapped market potential for lysosomal storage diseases is greater than $5 billion. Novazyme is well-positioned to move its technologies rapidly forward into the human clinic on multiple diseases and to take a leadership position in this therapeutic field.”
Series A 펀딩을 한지 딱 1년 후 9/11 테러가 난지 보름정도 지난 어수선한 상황에서 Genzyme이 Novazyme을 $206 Million에 인수하여 John Crowley는 Genzyme에서 Pompe Disease Program VP로 일하며 두명의 자녀들이 임상시험을 받게 하기 위해 애쓰다가 Conflict of interest 문제로 회사를 그만두게 됩니다.
Home-grown biotech company Novazyme officially has become part of international pharmaceutical company Genzyme General, company officials said Thursday.
The approximately $206 million acquisition was completed Wednesday with Oklahoma City-based Novazyme Pharmaceutical’s private group of stockholders getting about $19 million less than expected.
In August, a month before the Sept. 11 terrorist attacks that precipitated plunging stock prices, the companies announced the arrangement that was supposed to be worth about $225 million.
That figure was based on Genzyme’s initial payment of about 2.6 million shares of its stock to Novazyme’s shareholders. The stock closed at $56.90 that day. On Wednesday, when the deal was completed, the stock was more than $11 lower at $45.65.
The value of the deal is based on Genzyme General’s stock prices over the closing day and the previous three days. The value, including warrants, acquisition costs and stock purchase rights, combined with two upcoming payments totaling $87.5 million, is expected to be between $204 million and $209 million.
The lowered value was not surprising because Genzyme is among the many companies that have watched frightened investors dump their stock since Sept. 11.
Tulsa financial analyst Fred Russell said the declining value is not a great concern and the deal continues to be favorable.
“The difference between $225 million and $206 million, given the perspective of the last 60 days, is no decimation. We have two strong companies getting together and that should be good for everyone,” Russell said. “There’s nothing that has shown the fortunes of the two companies have changed.”
Genzyme expects the acquisition to enhance its position in the development of enzyme replacement treatment for lysosomal storage disorders. Novazyme founder and chief scientist Dr. William Canfield developed breakthrough technology to treat the disorders that include Pompe disease, a rare genetic disease that frequently kills children before they reach school age. Novazyme was approaching human clinical studies when Genzyme contacted Canfield and President and Chief Executive John Crowley about selling their company.
Genzyme’s plans call for the work to continue in Oklahoma City. Crowley said he expects the 70 local employees to increase to 100 within a year. After Genzyme completes the human trials and gets Federal Drug Administration approval to market the first two products in the United States, Novazyme shareholders stand to receive two additional payments totaling $87.5 million. Crowley hopes FDA approval will come in 2003.
“We are eager to start working closely with the Novazyme team to develop the best possible products for patients,” Jan van Heek, executive vice president of Genzyme Corp., said in a statement. “Novazyme’s technology platform, drug development team and first-class group of scientists will complement and significantly expand our product development capabilities.”
Canfield will continue to lead the scientific team at the Novazyme lab at 800 Research Parkway. Crowley will work as a senior vice president of Genzyme’s therapeutics unit, assuming overall responsibility for the Pompe disease programs. He also will continue as president of Novazyme, now a wholly owned subsidiary of the No. 3 ranked pharmaceutical company, Genzyme.
The acquisition should lower Genzyme’s earnings per share by 3 cents for 2001. Genzyme expects earnings of about $1.12 to $1.17 per share for the year, compared with previous estimates of $1.15 to $1.20 per share because of the issuance of the new shares for the acquisition.
Genzyme General stock closed up 2 percent Thursday at $46.60.
이러한 John Crowley의 노력에 힘입어 Novazyme을 설립한지 6년만인 2006년 Myozyme이 FDA 승인을 받을 수 있게 되었습니다. Myozyme이 승인되어 Pompe Disease를 치료할 수 있게 되었지만 이 약물은 Enzyme Replacement Therapy의 정상적인 Enzyme folding이 오래 지속되지 못하는 문제로 인해 질병 치료에 한계를 드러냅니다. 따라서 John Crowley는 이 문제를 해결하기 위해 두번째 회사인 Amicus Therapeutics를 창업하게 됩니다.
Genzyme Corp. announced today that the Food and Drug Administration has granted marketing approval for Myozyme(R) (alglucosidase alfa) in the United States. Myozyme has been approved for the treatment of patients with Pompe disease, a debilitating, progressive and often fatal disorder affecting fewer than 10,000 people worldwide. The product is the first treatment ever approved for Pompe disease and the first for an inherited muscle disorder.
“This is a special day for people across the Pompe community and at Genzyme, who have worked together for many years and overcome enormous challenges so that patients with this devastating disease now have a chance,” said Henri A. Termeer, chairman and chief executive officer of Genzyme Corp.
The Myozyme label includes the following indication: “Myozyme (alglucosidase alfa) is indicated for use in patients with Pompe disease (GAA deficiency). Myozyme has been shown to improve ventilator-free survival in patients with infantile-onset Pompe disease as compared to an untreated historical control, whereas use of Myozyme in patients with other forms of Pompe disease has not been adequately studied to assure safety and efficacy.”
The product label also includes a boxed warning with information on the potential risk of hypersensitivity reactions associated with Myozyme. The boxed warning states that “Life-threatening anaphylactic reactions, including anaphylactic shock, have been observed in patients during Myozyme infusion. Because of the potential for severe infusion reactions, appropriate medical support measures should be available when Myozyme is administered.” Of the 280 patients who received Myozyme in clinical studies or through expanded access, eight patients (3 percent) experienced severe or significant hypersensitivity reactions. Full prescribing information for the product is available on Genzyme’s Web site: http://www.genzyme.com/components/highlights/mz_pi.pdf
Pompe disease manifests as a broad spectrum of clinical symptoms. All patients typically experience progressive muscle weakness and breathing difficulty, but the rate of disease progression can vary widely depending on the age of onset and the extent of organ involvement. When symptoms appear within a few months of birth, babies frequently display a markedly enlarged heart and die within the first year of life. When symptoms appear during childhood, adolescence or adulthood, patients may experience steadily progressive debilitation and premature mortality due to respiratory failure. They often require mechanical ventilation to assist with breathing and wheelchairs to assist with mobility.
Genzyme recently completed enrollment in its clinical trial involving patients with late-onset Pompe disease. Ninety patients have been enrolled in this international, placebo-controlled study. Currently, more than 280 patients in 30 countries are receiving Myozyme through clinical trials, expanded access programs, or pre-approval regulatory mechanisms.
Myozyme has received orphan drug designation in the United States, which provides seven years of market exclusivity. The orphan drug law is designed to encourage the development of treatments for rare disorders such as Pompe disease, for which no therapies have existed previously. Genzyme expects to launch Myozyme in the United States within two weeks. Late last month, Myozyme was approved in the European Union.
Because early diagnosis, intervention and treatment are critical in Pompe disease and other lysosomal storage disorders, Genzyme has for the past seven years supported several outside research collaborations to develop new diagnostic technology. This research has led to the recent introduction of an enzyme assay utilizing blood samples that makes it possible to diagnose Pompe patients more rapidly and with a less-invasive procedure. Genzyme will offer this test now through its Genzyme Genetics unit, and the test will also be available through several other clinical laboratories in the United States and elsewhere in the world.
“The journey from development to approval of a therapy for Pompe disease has been a long and winding road, but we are now at a milestone and are thrilled with the outcome,” said Randall H. House, chairman of the International Pompe Association and president of the Acid Maltase Deficiency Association (AMDA), a Pompe patient association in the United States. “Enzyme replacement therapy with Myozyme gives Pompe patients hope.” The AMDA, formed in 1995, has assisted in funding Pompe disease research and promotes public awareness of Pompe disease.
Valerie Cwik, medical director for the Muscular Dystrophy Association, said: “Myozyme is the first treatment for any of the muscle diseases included among the 40 neuromuscular disorders covered by the Muscular Dystrophy Association. This is a great day for people with Pompe disease, and a hopeful moment for the thousands of other people who are affected by the diseases in the MDA program, because it shows that support and research can lead to treatments.” The MDA helped support patients who took part in clinical trials of Myozyme and also sponsored early research in Pompe disease.
Genzyme began working to develop a treatment for Pompe disease in 1998. In 2003, the company initiated a pivotal clinical study of Myozyme that demonstrated the product’s safety and efficacy. In the study, 83 percent of patients treated with Myozyme were both alive and free of invasive ventilator support at 18 months of age. In a natural history study, 2 percent of similar infantile-onset patients were alive at 18 months of age. The pivotal trial enrolled 18 patients with infantile-onset Pompe disease, who began receiving therapy at approximately six months of age. The most common serious adverse events observed in clinical studies of Myozyme, whether or not they were related to the drug, were pneumonia, respiratory failure, respiratory distress, catheter-related infection, respiratory syncytial virus infection, gastroenteritis and fever. Many of these can be complications of Pompe disease.
“We are very proud that we have been able to bring to market four therapies for ultra-orphan diseases where no treatments existed previously,” said Mr. Termeer. “This underscores our fundamental commitment to patients and confirms the productivity of our research efforts. We continue to invest in potential new approaches to treating these diseases.”
Myozyme is the fourth enzyme replacement therapy developed by Genzyme for a rare genetic disease. Genzyme has developed Cerezyme(R) (imiglucerase for injection) for Type 1 Gaucher disease; Fabrazyme(R) (agalsidase beta) for Fabry disease; and, in collaboration with BioMarin Pharmaceutical Inc., Aldurazyme(R) (laronidase) for MPS I. These treatments are currently available to patients throughout the world.
Genzyme currently manufactures Myozyme in the United States. In the future, the company expects to also produce Myozyme at its new protein manufacturing facility in Geel, Belgium, and its new fill/finish facility in Waterford, Ireland, to ensure that it is able to meet the anticipated demand for the product throughout the world.
About Pompe Disease
Pompe disease, also known as Acid Maltase Deficiency or Glycogen Storage Disease Type II, is one of more than 40 genetic diseases called lysosomal storage disorders, which are caused by a deficiency or malfunction of specific enzymes found in cell lysosomes. People born with Pompe disease have an inherited deficiency of an enzyme known as acid alpha-glucosidase (GAA). Enzymes, which are protein molecules within cells, trigger biochemical reactions in the body. In a healthy person with normal GAA activity, this particular enzyme would assist in the breakdown of glycogen, a complex sugar molecule stored within a compartment of the cell known as the lysosome. But in Pompe disease, the GAA activity may be dramatically reduced, dysfunctional, or non-existent, resulting in an excessive accumulation of glycogen in the lysosome.
Eventually, the lysosome may become so clogged with glycogen that normal cellular function is disrupted and muscle function is impaired. Although there is glycogen storage in the cells of multiple tissues, heart and skeletal muscles are usually the most seriously affected.
Amicus Therapeutics는 Myozyme에서 경험한 Enzyme folding 문제를 해결하기 위해 초기부터 Chaperone 작용제인 경구용 화학요법제를 개발하기로 합니다. 그리고 2004년에 $31 Million Series B를 합니다. Amicus는 Pompe Disease 뿐만 아니라 같은 문제를 가진 Fabry나 Gaucher 와 같은 병을 함께 치료하는 더 큰 과제를 수행하기 위해 노력했고 Series B를 받은 당시 가장 먼저 진행하던 약물은 Fabry 치료제인 AT1001로서 전임상을 진행 중에 있었습니다.
Amicus Therapeutics, Inc., an emerging drug development company focused on the development of a novel therapeutic approach to the treatment of human genetic disorders, with an initial focus on lysosomal storage diseases, today announced the completion of a $31 million Series B private equity financing. The Series B Round was led by Canaan Partners, L.P., with participation from other new investors, Frazier Healthcare Ventures, L.P., New Enterprise Associates, L.P., Prospect Venture Partners, L.P., and Radius Venture Partners, L.P. The company’s founding investor, CHL Medical Partners, also participated in the round.
“Collectively, these investors have an excellent track record in helping to build significant and successful companies, and they bring considerable experience and knowledge to the table in addition to financial resources,” said Norman Hardman, Ph.D., Chief Executive Officer of Amicus Therapeutics, Inc. “With their commitment and support, Amicus is firmly on its way to realizing its vision of becoming the premier company developing treatments for human genetic diseases with small-molecule drugs. During the fundraising we were able to stay focused on ensuring that the pre-clinical development of AT1001, our lead product candidate for treatment of Fabry disease, remained on track. The progress we made has clearly impressed our investors, all of whom are extremely excited about this new product opportunity. With this round of financing secured, our top priorities will be the further advancement of AT1001 –which we plan to have in the clinic by the third quarter of this year — and the development of our R&D program for Gaucher Disease.”
With the close of the financing, Stephen Bloch, M.D., of Canaan Partners, James Topper, M.D., Ph.D., of Frazier Healthcare Ventures, Mike Raab of New Enterprise Associates, and Alex Barkas, Ph.D., of Prospect Venture Partners, have joined the board. They join existing board members, Norman Hardman , Ph.D., and Gregory Weinhoff, M.D., of CHL Medical Partners.
“Successfully concluding this phase of our corporate development brings us a significant step closer to providing potentially effective and convenient oral therapies to those who suffer from Fabry disease, Gaucher disease, and a range of other genetic disorders,” concluded Dr. Hardman.
1년만에 Fabry 치료제 Amigal *migalastat hydrochloride)는 임상2상까지 진행시킬 수 있었고 임상시험 진행과 다른 파이프라인 개발을 위해 $55 Million Series C Funding을 합니다. Gaucher 치료제인 AT2101도 전임상 단계에 이미 진입한 상태였습니다.
Amicus Therapeutics, a biopharmaceutical company developing small molecule, orally-active pharmacological chaperones for the treatment of human genetic diseases, today announced the closing of a $55 million Series C financing. The company intends to use the proceeds to advance its drug pipeline based on the company’s unique technology that has the potential to transform the treatment of human genetic diseases. Amicus’ lead compound Amigal (migalastat hydrochloride) is in a Phase ll clinical program for Fabry disease. All of Amicus’ current investors participated in the Series C round, which was led by new investor Quaker BioVentures.
“Amicus has truly exciting technology that has breakthrough potential for the treatment of devastating genetic diseases,” said Sherrill Neff, managing partner of Quaker BioVentures. “We welcomed the opportunity to join Amicus’ current investors and lead this financing as the company continues to advance. Amicus will soon have two promising compounds in clinical trials based on its pharmacological chaperone technology, and with this new financing the company is well positioned to continue to ramp up its multiple drug development activities.“
Quaker BioVentures led the Series C financing, joined by existing investors Canaan Partners, CHL Medical Partners, Frazier Healthcare Ventures, New Enterprise Associates, Prospect Venture Partners and Radius Ventures. Other new investors include Palo Alto Investors and the Garden State Life Sciences Venture Fund, which is funded by the New Jersey Economic Development Authority (NJEDA) and managed by Quaker BioVentures. Mr. Neff of Quaker BioVentures is joining the Amicus Board of Directors.
“As an early and now repeat investor in Amicus, we are most pleased and impressed with the company’s achievement of several significant milestones in just the past nine months. The momentum at Amicus is simply remarkable and reflects the breadth and depth of its core technology as well as the commitment and passion of its now 35 employees,” said Michael Raab, a partner at New Enterprise Associates and an Amicus board member.
“At Amicus, we are building momentum in human genetic diseases,” said John F. Crowley, chairman and chief executive officer of Amicus. “We look forward to applying these new financial resources to advance our lead compound, Amigal, and to accelerate the growing momentum of our preclinical programs. We are optimistic that our passion and commitment to this field will rapidly translate into effective therapies for the many individuals who live with these life threatening disorders.”
Separately, Amicus today announced positive results from its Phase l clinical studies of Amigal and the start of patient enrollment in its Phase ll clinical program for the treatment of Fabry disease. In addition to meeting all of its safety endpoints, the Phase l studies demonstrated proof of concept for Amicus’ pharmacological chaperone technology, showing that Amigal has the ability to increase target enzyme activity levels, even in healthy individuals.
Amicus’ second product candidate, AT2101, a treatment for Gaucher disease, is in late stage preclinical development with clinical trials expected to commence in the first half of 2006. The company also is advancing its pipeline of earlier stage pharmacological chaperone compounds for a variety of human genetic disorders.
Amicus recently moved into a new 40,000 square foot, state-of-the-art business and science headquarters facility in Cranbury, NJ.
“Amicus represents another success story for the thriving New Jersey biotechnology sector and we are very pleased the Garden State Life Sciences Venture Fund is participating in this financing,” said Caren Franzini, chief executive officer of the NJEDA. “Amicus is a graduate of the EDA’s Commercialization Center for Innovative Technologies in North Brunswick, and we believe the company epitomizes the innovation, entrepreneurial savvy and industry expertise that are making our state such an attractive location for this critically important sector.”
이듬해에 Nasdaq IPO를 하려고 S-1 Filing을 했지만 시장 상황이 좋지 않은 관계로 IPO 를 포기하고 $60 Million Series D를 하게 됩니다. NEA의 주도로 기존 투자자들이 참여한 펀딩 라운드인데 기존 투자자들은 Amicus의 빠른 개발 속도와 실행력에 지원을 아끼지 않았습니다.
Amicus Therapeutics, a biopharmaceutical company developing small molecule, orally-administered pharmacological chaperones for the treatment of a range of human genetic diseases, today announced the closing of a $60 million Series D financing, the Company’s largest to date, led by New Enterprise Associates (NEA). In August, due to market conditions, the Company withdrew its S-1 registration statement with the Securities and Exchange Commission (SEC) relating to a proposed initial public offering.
In this financing round, NEA was joined by current investors Canaan Partners, CHL Medical Partners, Frazier Healthcare Ventures, Palo Alto Investors, Prospect Venture Partners, Quaker BioVentures, and Radius Ventures. In addition, affiliated investment funds of Och-Ziff Capital Management Group participated in this round of financing as a new investor.
Amicus also announced today two key executive management appointments. First, Donald J. Hayden, Jr., Executive Chairman, will serve as Interim President and Chief Executive Officer while John F. Crowley, President and CEO, serves a six-month active duty military obligation with the United States Navy. He is a commissioned officer in the Navy Reserve. Mr. Crowley will return as the CEO on or about March 1st, 2007. Second, Matthew R. Patterson has been promoted from Chief Business Officer to Chief Operating Officer.
“I am delighted that Amicus has been able to secure our largest financing round to date. Our core investors clearly believe in the Company’s pharmacological chaperone technology. We are also extremely pleased to have Och-Ziff Capital Management Group onboard as our newest investor. This additional funding will enable us to continue to advance our pipeline of potential new treatments for the lysosomal storage disorders Fabry, Gaucher, and Pompe and to aggressively develop our earlier stage programs for other genetic diseases. Additionally, I am honored to be asked to serve my country during this active duty obligation and I have the utmost confidence that Don Hayden and the extraordinary senior management team at Amicus will continue to move the company forward while I am away,” said John F. Crowley.
“We are extremely pleased to make this additional investment that will further enable Amicus’ dynamic growth and lay the foundation for a significant and enduring enterprise. The speed and high quality of execution are a testament to the outstanding efforts of this well led and capable team. We wish John Crowley the best of luck during his six-month military service and look forward to his return next spring,” said Michael G. Raab, Partner at NEA and member of Amicus’ Board of Directors.
Amicus Therapeutics (Nasdaq: FOLD) announced today the pricing of its initial public offering (IPO) of 5,000,000 shares of its common stock at a public offering price of $15.00 per share. Amicus has granted the underwriters a 30-day option to purchase up to an additional 750,000 shares of common stock to cover over- allotments, if any. All shares in the offering will be sold by the Company and are expected to begin trading today on the NASDAQ Global Market under the trading symbol “FOLD.”
IPO한지 반년이 지난 후 Shire가 $50 Million upfront를 포함한 총 $440 Million규모의 계약을 합니다. Chaperone technology에 대한 관심과 Amicus의 세가지 약물 Phase II Amigal™ for Fabry disease, Phase II Plicera™ for Gaucher disease, 그리고 Phase I AT2220 for Pompe disease 치료제에 투자합니다.
Shire is expanding its pipeline of therapeutics for rare genetic diseases through a licensing deal with Amicus Therapeutics that could be worth up to $440 million. The three Amicus candidates are based on chaperone technology, which will augment Shire’s enzyme-replacement therapies.
Shire will pay $50 million upfront for three compounds in markets outside of the United States: Phase II Amigal™ for Fabry disease, Phase II Plicera™ for Gaucher disease, and Phase I AT2220 for Pompe disease.
Phase II data for Amigal and preliminary Phase II results for Plicera will be available by year end, according to Matt Patterson, COO at Amicus. For AT2220, the firm plans to start a Phase II study early in 2008, he adds.
Based on development achievements through to approval of these compounds, Patterson says that Amicus is eligible to receive $150 million. The company may also get another $240 million in sales milestones. Shire will also make tiered, double-digit royalty payments on net sales.
The companies will jointly pursue a development program toward market approval in the U.S. and Europe and share expenses equally.
The pharmacological chaperone technology, which the firms expect to make available as oral therapies, has been applied to various enzymes that are defective as a result of improper folding. In contrast to the traditional enzyme-replacement approach, pharmacological chaperone technology involves the use of small molecules that selectively bind to and stabilize proteins in cells. This reportedly leads to improved protein folding and trafficking as well as increased activity.
“Amicus’ pharmacological chaperone compounds have the potential to be an excellent addition to our current enzyme-replacement therapy business,” states Shire CEO, Matthew Emmens. Shire’s pipeline includes enzyme-replacement treatments Replagal™ for Fabry disease and GA-GCB in Phase III development for Gaucher disease. “In addition,” continues Emmens, “it provides an opportunity for Shire to enter the market for Pompe disease.”
2010년에는 GSK가 임상3상이 진행 중이던 Fabry 치료제인 Amigal의 상업판권을 위해 upfront $30 Million, 지분투자 $30 Million을 포함한 총 규모 $170 Million의 계약을 합니다.
GlaxoSmithKline (GSK) and Amicus Therapeutics (Amicus) today announced a definitive agreement to develop and commercialise AmigalTM (migalastat HCl), currently in Phase 3 for the treatment of Fabry disease, a rare inherited disorder. Under the terms of the agreement, GSK will receive an exclusive worldwide license to develop, manufacture and commercialise migalastat HCl. Additionally, as part of the agreement, GSK and Amicus also intend to advance clinical studies exploring the co-administration of migalastat HCl with enzyme replacement therapy (ERT) for the treatment of Fabry disease.
Under the terms of the agreement, Amicus will receive an upfront license payment of $30M from GSK and is eligible to receive further payments of approximately $170M upon the successful achievement of development and commercialisation milestones, as well as tiered double-digit royalties on global sales of migalastat HCl. GSK and Amicus will jointly fund development costs in accordance with an agreed upon development plan. Additionally, as part of the collaboration, GSK is purchasing 6.9 million shares of Amicus common stock at a price of $4.56 per share. The total value of this equity investment to Amicus is $31 million and represents a 19.9% ownership position for GSK in the Company. The total cash upfront to Amicus from GSK for the license payment and equity investment is approximately $60 million.
“This strategic collaboration is another significant milestone in delivering our vision for GSK Rare Diseases. Amicus’ scientific and clinical expertise in human genetic diseases is among the best in the industry, and we are pleased to be collaborators and investors in this exceptional company” said Marc Dunoyer, Global Head of GSK Rare Diseases. “Our focus now is to continue to advance Amigal for Fabry disease and it is our hope to deliver a first-in-class, oral medicine to the thousands of people worldwide living with this devastating rare disease.”
John F. Crowley, Chairman and Chief Executive Officer of Amicus Therapeutics said, “The completion of this agreement with GSK is a transformational event for Amicus. It provides a strong validation of the potential for Amigal to become an important new treatment option for people living with Fabry disease and for our pharmacological chaperone technology broadly. GSK has extremely impressive global clinical, regulatory and commercial expertise and a strong commitment to the development of treatments for rare diseases. We look forward to working in close partnership with them.” Crowley continued, “With this key strategic alliance with GSK and the added financial strength it provides, Amicus is now uniquely positioned to build shareholder value through our expertise in rare disease drug development.”
2013년에는 Biogen이 Parkinson 치료를 위한 표적인 lysosomal enzyme glucocerobrosidase (GCase) enzyme 공동개발 협약을 맺습니다. 금액은 공개하지 않았으나 모든 연구비와 활동비를 Reimbursement 하는 계약이었습니다.
Amicus Therapeutics said today it will partner with Biogen Idec to discover small molecules that fight Parkinson’s disease by targeting the lysosomal enzyme glucocerobrosidase (GCase) enzyme, with the biotech giant overseeing their further development and commercialization. The value of the multiyear collaboration was not disclosed.
Biogen Idec agreed to fund all discovery, development, and commercialization activities, as well as reimburse Amicus for all full-time employees working on the project. Amicus is also eligible to receive from Biogen Idec payments based on undisclosed development and regulatory milestones, as well as what it termed “modest” royalties on global net sales.
Amicus는 NDA rolling submission 중이던 epidermolysis bullosa라는 피부 희귀질환치료제Zorblisa (SD-101)를 개발하는 Scioderm을 $229 Million 주식교환 및 $618 Million milestone payment, 그리고 PRV voucher 를 인수하는 계약을 함으로써 새로운 파이프라인을 얻습니다. 이 계약은 지난 2년간 Scioderm의 이사회 멤버로 약의 개발과정을 지켜본 John Crowley의 결정이었습니다.
Amicus Therapeutics has stepped in to snap up the late-stage rare disease biotech Scioderm, beefing up its orphan drug pipeline in exchange for $229 million in stock and cash along with a promise of up to $618 million more for meeting a slate of milestones.
The buyout leaves Amicus ($FOLD) with its lead drug Galafold under regulatory review, a new drug that could be filed in the near term and a third program entering the clinic–with CEO John Crowley prepping for more deals as the company builds out a global commercial team.
Amicus completes this buyout a little more than a month after Scioderm started a rolling submission of its NDA for Zorblisa (SD-101), an experimental drug for epidermolysis bullosa, a condition that leaves children’s skin papery thin and fragile, subject to tearing and blistering. Most patients–and there are some 30,000 to 40,000 in the world’s major drug markets–die before the age of 30.
Data from the late-stage study is due in the first half of next year, with Amicus looking for a regulatory green light on a drug Crowley believes could be a blockbuster. The market for Zorblisa, says the CEO, is worth “a billion dollars-plus.” Adds Crowley: “We want to be one of the leading biotech companies focused on rare diseases.”
Not that the deal is without risk. TheStreet notes that Zorblisa failed its Phase IIb study, with a small subgroup of patients in the top dose achieving statistical significance compared to a placebo. That top dose is being taken into the Phase III with the blessing of regulators.
There’s an added bonus involved in this deal. If Zorblisa is approved, the owners could qualify for a priority review voucher–an asset that’s been worth hundreds of millions of dollars in recent deals. Amicus will pay Scioderm’s investors either $100 million or half the sales price for the voucher, whichever is less.
Crowley has had a front row seat on Scioderm’s progress as a board member of the company for the last two years.
Little Durham, NC-based Scioderm was unique among the virtual crowd back in 2013, when the FDA handed out one of its first breakthrough therapeutic designations to the biotech. The company raised a $20 million B round late last year and was named a Fierce 15 company back in 2013, when it was still in early development. At the time of the buyout, its staff had grown to 9 and CEO Robert Ryan will now join Amicus in a senior executive position.
Amicus has been focused primarily on Galafold (migalastat), a treatment for Fabry disease. The biotech experienced a severe setback a few years ago when the drug failed a comparison study with a placebo. The failure spurred GlaxoSmithKline ($GSK) to drop out of its partnership, but Amicus came back with a new plan to use a different biomarker on symptoms of the disease. That strategy paid off with successful Phase III studies of the drug, comparing well with Sanofi’s ($SNY) Fabrazyme and Shire’s ($SHPG) Replagal in two measures of kidney function. And that should help make the case for switching patients from a regularly infused drug that costs more than $200,000 a year to an oral therapy.
European regulators have granted an accelerated review of the treatment while an NDA at the FDA is slated for delivery before the end of this year. Amicus has hired 20 people for its European commercial operation, says the CEO. As new products are added, he forecasts, that should grow into a global operation with hundreds of staffers pursuing sales of $500 million to more than a billion dollars on each of three products. As this deal cost $125 million in cash, he says Amicus’s business development team is well positioned to hunt down more tech and product deals, with an early focus on some early-stage deals this time around.
Amicus is hunting new deals in a hot market. Rare disease therapies can earn well into 6 figures, making it an attractive market for a host of companies. Other companies like Alexion ($ALXN) and BioMarin ($BMRN) have done extremely well with investors looking to share the profits. Crowley has a personal connection to the genetic diseases he focuses on. In 1998, two of his children were diagnosed with Pompe disease.
작년 9월 마침내 Pompe 치료제인 “Pombiliti (cipaglucosidase alfa-atga) + Opfolda (miglustat)” 컴비네이션 약물이 FDA승인을 받게 됩니다. 실로 Novazyme을 설립한지 20여년이 걸린 오랜 기간의 노력이었습니다.
Amicus Therapeutics has received approval from the US Food and Drug Administration (FDA) for a two-component therapy, Pombiliti (cipaglucosidase alfa-atga) + Opfolda (miglustat) 65mg capsules, to treat Pompe disease. Pombiliti treats adult patients with late-onset Pompe disease (LOPD) weighing 40kg or under who are not able to improve on their present enzyme replacement therapy (ERT). LOPD is a lysosomal disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA).
Designed for enhanced uptake into muscle cells, pombiliti is a recombinant human GAA enzyme (rhGAA) naturally expressed with high levels of Mannose 6-Phosphate (bis-M6P).Upon entering the cell, it will be processed into the active and mature form to break down glycogen. The FDA has approved the therapy based on clinical data from the Phase III PROPEL pivotal study. It is the only LOPD trial to assess ERT-experienced participants in a controlled setting. Amicus Therapeutics president and CEO Bradley Campbell stated: “Today’s approval is also a testament to Team Amicus’ extraordinary dedication to patients and our ability to execute on our vision to bring new therapies to the rare disease community.
“Our highly experienced team is ready to launch this medicine in the US and we look forward to rapidly bringing this new treatment regimen to all eligible adults living with late-onset Pompe disease who are not improving on their current ERT.”
Amicus의 역사는 여기서 끝이 아닙니다. Pompe 치료제 승인과 함께 $30 Million 지분투자 및 $400 Million Loan을 받는 계약을 통해 상업적 성공을 위해 노력하고 있습니다.
Hot off an FDA approval for its Pompe disease combo treatment, Amicus Therapeutics has reeled in a major investor. Monday, Amicus unveiled a $430 million financing pact with Blackstone Life Sciences and Blackstone Credit. The deal will see the asset manager furnish Amicus with a $400 million loan that Amicus will use to refinance existing debt, Amicus said in a release. Additionally, Blackstone made a $30 million strategic investment in Amicus’ common stock. The move is designed to help Amicus grow revenues and move toward profitability, the company said.
Late last month, Amicus notched a big win with the FDA approval of Pombility and Opfolda. The green light covers adults living with late-onset Pompe disease who aren’t improving with current enzyme replacement therapy (ERT). Pombiliti is an infused long-term ERT, while Opfolda is an oral stabilizer, which reduces the loss of enzyme activity in the blood during infusion.
The company figures the Pompe disease market could reach $1.8 billion by 2027 and projects peak sales of its combo will reach $1.2 billion. Last week, the company said it was ready to launch “immediately” in the U.S., where it has priced the treatment at $650,000 per year for a patient of medium weight.
Aside from its new Pompe combo, Amicus boasts another commercial product in Fabry disease treatment Galafold, which won its U.S. nod in August of 2018. Amicus’ full-year revenues for 2022 clocked in at $329.2 million, a year-over-year increase of 8%. In March, the company telegraphed a potential pivot to profitability by the second half of 2023.
Pompe 치료제의 FDA 승인에 결정적 임상이었던 PROPEL의 임상결과는 아래의 Presentation에 잘 나타나 있습니다. John Crowley의 노력으로 Pompe 치료제로 고생하는 두자녀를 포함한 세자녀와 아내, 총 다섯식구는 지금도 행복한 가정을 이루고 살고 있습니다. 또한 John Crowley는 신약개발자의 롤모델로서 2024년도 BIO의 대표 (President & CEO)로 선출되는 영예도 얻었습니다. John Crowley의 삶은 신약개발자가 어떤 마음으로 신약을 개발해야 하는지 잘 보여준 살아있는 표본이라고 할 수 있습니다. 그는 현재 Amicus Therapeutics의 Executive Chairman으로 회사를 이끌고 있습니다.
Brown University의 생물공학부 학부생이었던 Josh Cohen과 Justin Klee는 연구논문을 뒤지다가 Neuronal Cell Death에 대해 공부를 하던 중 Phenylbuturate (PB)와 Tauroursodeoxycholic acid (TUDCA)가 각각 효과를 가지고 있다는 것을 발견한다. 아래의 두가지 논문이 대표적으로 이 각각의 약물의 효과를 증명하였다.
그들은 PB-TUDCA combo를 통해서 Neuronal cell death를 막을 수 있다면 Neurodegenerative disease를 치료할 수 있지 않을까? 하는 단순한 아이디어를 가지고 Preclinical test를 하여 PB-TUDCA Combo Hypothesis를 증명한다. 아래 논문이 그 결과를 보여준다.
Amylyx Pharmaceuticals를 설립한다. 경험이 없는 이들에게 Angus Kingon1 교수가 특허출원을 조언하고 Jonathan M. Nelson의 멘토링을 통해 Startup grant와 ALS grant를 확보하고 Preclinical과 임상시험 시작을 할 수 있게 되고 $5 Million Series A을 통해 임상2상을 시작할 수 있게 된다. 임상 2상 CENTAIR 결과를 통해서 FDA 승인을 얻고 판매를 시작했으나 최근 임상3상 데이타가 부정적으로 나오면서 ALS 치료제로 지속할 수 없는 위기에 처하게 되었다. 하지만 아직도 희귀질환 뇌질환 두가지에 대한 임상3상이 계속 진행할 예정이므로 AMX0035가 어려움을 겪고 있지만 약 자체가 실패했다고 볼 수는 없을 것 같다. 기대를 가지고 지켜봐야할 것 같다.
Biomedical engineering alumnus Josh Cohen ’14 partnered with Justin Klee ’13 eight years ago to build a company dedicated to the development of therapeutics for the treatment of neurodegenerative disorders.
In the Cambridge, Mass. conference room where the results of the CENTAUR clinical trial for AMX0035 were delivered to co-founders Josh Cohen and Justin Klee, the excitement was palpable. The study showed that Amylyx Pharmaceuticals, Inc.’s amyotrophic lateral sclerosis (ALS) pipeline drug showed potential to prolong patient survival and slow rapid disease progression in ALS patients, analyzing 137 patients enrolled at 25 top ALS medical centers across the United States.
The functional and survival benefits of AMX0035 significantly strengthened the drug’s position to enter the ALS market over the coming years as a potential disease-modifying therapy. “But the point where it really hit us was when we talked to a nurse at Mass General Hospital in the days following,” Cohen said. “The nurse said, ‘Every day I have to tell people that their life is being cut substantially short. There are a lot of tears in clinic. But today I was able to share a ton of hope. It was a different day.’
“Things like that are why you do it. The patients are why we get out of bed in the morning.” Cohen said, explaining how the patients who participate in Amylyx trials are the motivation behind the company’s progress. “There was a time we were having a significant issue with the manufacturing process, and there just seemed no way to do it. It was a pretty big hold up, and we were in a stage where we had investors, had built up hope, and were not sure of a path forward. After a two-hour meeting with a patient, who was at a stage where he was speaking through an eye tracker but still found joy and laughter, we walked out with a different perspective of what people with ALS were dealing with on a daily basis. We found a way through the issue.”
Klee and Cohen were recent guests on the Brown University Carney Institute for Brain Science’s “Carney Conversations,” where Klee recounted the genesis of Amylyx. “We had been friends since Josh’s first year, and one day he shows up after I hadn’t seen him in long time, and said, ‘I’ve been reading all these papers and thinking about Alzheimer’s and neurodegeneration and I think I may have a way of looking at it and treating it.’”
Then a junior, Cohen was in the literature-heavy portion of his studies. “I just got really into that,” Cohen said. “This idea that there are literally millions of papers out there that are waiting to be read, and integrated – I ended up reading a lot, particularly about neuronal death, trying to look at it in a way that there might be more to try.
“The literature search was really my own, but courses that were making us read a lot at that time were a stem cell engineering course, and Biomaterials with Tayhas Palmore. I took Neuroengineering with Leigh Hochberg, and then my pharmaceuticals interest came from Drug and Gene Delivery with Edith Mathiowitz, and also a Physiological Pharmacology course. Once we started Amylyx, it became my capstone with Dr. (Anubhav) Tripathi.”
Klee was intrigued with Cohen’s early theories, and scientific curiosity led the way for starting the company. “These are such enormous problems,” said Klee. “What if we found a way to treat them that people haven’t yet thought of? This is such an amazing industry. It’s scientifically interesting from both a business and people perspective. And at the end of day, if we’re successful, we’ve helped people who really need it,” he said. “So we kept reading and the short story is, we said ‘Let’s start a company.’ In retrospect, we had no idea what that meant.”
Cohen said Professor Angus Kingon was the engineering adviser the pair leaned on most heavily, and was one of the first people to encourage and offer advice on securing a patent. Alan Harlam, founding director of the Social Innovation Initiative (SII) at Brown’s Swearer Center, and now mentor in the Jonathan M. Nelson Center for Entrepreneurship, was another early adviser of Amylyx. A startup grant from Swearer helped Cohen and Klee scrap together enough money to run the first experiments at Charles River Labs.
“It may look from the outside that Justin and I drove everything, but the truth is every step of the way so many advisers and different people were giving us tips and tricks on how to get to the next stage. Brown is such a great place to be for it with its entrepreneurial spirit, starting with the open curriculum, encouraging self-directed thinking,” said Cohen.
“We had the advantage of not knowing, not having a full appreciation of what it might take to do this. Once we had fallen in love with the idea, we thought if we didn’t pursue it, it wouldn’t be pursued. It wasn’t easy: Inexperienced leaders going after diseases that had historically been impossible, with a project that wasn’t going to be cheap. There were many times we wondered if there was a path forward. A few advisers took a chance on us, and told us to keep pushing. That helped a lot.
“The nice thing about doing this at the undergrad age is if the thing totally fails, it’s not as bad as it could be. If I could boil the last eight years into a few sentences, it would include preparing for unexpected challenges. As we were developing, the hardest thing was manufacturing. The standards that are expected for pharmaceutical manufacturing is a tightrope. And it requires a lot of iteration and a lot of frustration,” Cohen said.
“One summary of the whole process is get a little data, raise a little money, get a little more data, raise a little more money. It makes the whole process very stressful and difficult, because you end up fundraising quite frequently and it’s never far away that you’ll have to be fundraising again,” he said. “One thing that was really transformative for us, one uphill battle we fought, was our credibility, because we are young founders. We had to overcome that, and one of the biggest helps for that was a grant from the ALS Association that helped support the clinical trial. Both the money and implied credibility of that was helpful.
“I’ve always felt Brown doesn’t get enough credit on the entrepreneurship side. The school almost naturally recruits entrepreneurs. The open curriculum means you can take whatever you want, so Brown students take that in all sorts of directions. I mean, there are some whose schedule looks totally indistinguishable from if they went to another school, and there are students who totally focus in one area or take one of everything.
“It naturally selects kids who want to be self-directed, self-motivated, who want to build their own thing, brand, skillset and persona. It’s really exciting what Brown is doing with the Nelson Center to bring that out and operationalize that. One of the things I really appreciate about Brown engineering is the mentality of self-direction. Many of the early classes, and certainly in the later classes, professors are likely to give you a couple of tools and encourage you to figure it out. That can be frustrating, but I loved it. I thought it made you re-think how you approach problems. I still find everything I learned from Brown Engineering incredibly helpful at Amylyx, but especially the problem solving. It’s a hard program, so when you’re done with it, you sort of know how to manage your life, your schedule, and your time. We’ve had a couple of candidates come to interview who are Brown engineers and I always say, ‘Let’s put them at the top of the list.’”
The co-CEO business model, considered by many an unusual way to structure a company, seems to work advantageously for Klee and Cohen. “It helps a lot that we are very good friends,” Klee said. “We have a fundamental trust that we both have the same vision, but we usually approach problems completely differently, me from neuroscience and Josh from biomedical engineering. But I know when Josh comes forward, it is going to be well thought out and something I haven’t thought about. We recognize our strengths and weaknesses and how we each have our different way of doing things. Most big problems you want to talk it through and have a debate, anyway. On the smaller issues, we have decision makers at different places at the same time.”
Cohen added, “Collaboration is everything. If your partners and advisers are unpleasant to work with you won’t get far. Two-way respect is what makes it work. And a lot comes down to self-awareness. None of us is perfect. We realized early what we’re good at, and what we’re bad at.”
With the most recent trial results showing such promise, Amylyx now looks to the future. “The CENTAUR trial showed that people with ALS could retain their functional abilities for more time and survive longer. With that data the question is, is that enough for approval and marketing, or are additional studies needed? We’ve decided to file for approval in Canada and Europe. In the United States, it seems likely we will need additional data. So we’re planning an additional study to support that,” Cohen said. “Our goal now for the company is marketing the drug, building operations and the infrastructure to support that while working to complete any necessary additional trials. We are focused on that right now. We really want to do an excellent launch on this drug candidate for ALS before we look too far past that,” he said.
“We are thrilled about AMX 0035. These are terribly hard diseases, so a significant outcome is awesome, but it is not a cure. As our company continues to grow, we feel we have a social contract to reinvest those resources into driving more and better solutions for neurodegenerative disease. ALS is obviously our focus, but we also have a trial ongoing in Alzheimer’s, and are researching other neurodegenerative diseases as well. This is definitely the space we want to stay in, where we’ve developed our relationships, and have our expertise.”
Amylyx Pharmaceuticals Inc. announced today that it has completed a $5 million Series A financing to support its upcoming Phase II trial in patients with Amyotrophic Lateral Sclerosis (ALS). Morningside Venture led the financing and was joined by the ALS Investment Fund and former Genzyme CEO Henri Termeer, as well as new and previous private investors.
The financing adds to a recently awarded $2.96 million grant to support the clinical trial from the ALS Accelerated Therapeutics Initiative, which is a collaboration between the ALS Association and ALS Finding a Cure. Overall, Amylyx has raised $10 million in grant funding and private financing to advance AMX0035. The IND for this Phase II trial is on schedule for the fourth quarter of 2016 and the trial will start shortly thereafter. Over 20 clinical sites across the United States have already expressed interest in participating.
In connection with the financing, Dr. Isaac Cheng, M.D. from Morningside and Mr. Felix von Coerper from the ALS Investment Fund have joined Amylyx’s board of directors. Mr. Termeer will become a Board observer. They will join directors Dr. George Milne, former President of Central Research and President of Strategic and Operations Management at Pfizer; Dr. Walter Gilbert, Nobel Laureate and former Biogen CEO; and Stephen D. Chubb, previous CEO of three publicly-traded biotechnology companies and previous chairman of the board at Mount Auburn Hospital.
“Amylyx has built a strong rationale for AMX0035 in the treatment of ALS and has developed an outstanding clinical plan to evaluate its potential in ALS patients,” said Dr. Isaac Cheng M.D. of Morningside. “There is a critical unmet need for ALS treatments and Morningside is pleased to support Amylyx’s program.”
“With AMX0035, Amylyx seeks to advance a promising approach to ALS therapy, by targeting neuroinflammation and cell death, that is now poised to enter human studies,” said Mr. von Coerper. “Our investment in Amylyx is aligned with the Fund’s goals of driving medical advancement for this terrible disease.”
“Amylyx’s clinical plan is not only designed to validate their therapeutic candidate, but also push forward important innovation in trial design and execution in ALS,” said Henri Termeer. “I am excited to work with Amylyx as they work to make a difference in the lives of these patients.”
AMX0035 combines two drugs, sodium phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA), that Amylyx has shown act synergistically in preclinical models of ALS. Previous studies with PB and TUDCA as single agents demonstrated efficacy in several cellular and animal models of ALS. In addition, PB and TUDCA have been individually tested in clinical trials of ALS and both showed safety and tolerability and preliminary signs of efficacy.
The Phase II clinical trial will test the safety and tolerability of AMX0035, as well as functional outcomes. Analysis of biomarkers of cell function, neuronal damage, and inflammation will be included as a major part of the trial, along with a new measure of muscle strength that has the potential to be an objective and sensitive assessment of disease progression.
“We are very pleased with the support and commitment of this group of highly experienced investors who share the focus of making a significant difference in this disease,” said Justin Klee, President at Amylyx. Josh Cohen, CEO added, “We are also thrilled to welcome our new Board members, and anticipate benefiting from their guidance as we prepare to enter clinical development with AMX0035.”
ALS is a progressive neurodegenerative disease that affects nerve cells in the brain and the spinal cord. Eventually, people with ALS lose the ability to initiate and control muscle movement, which leads to total paralysis and death, usually within two to five years of diagnosis. There is no cure, and only one drug approved by the U.S. Food and Drug Administration modestly extends survival.
A month after filing for Canadian approval for its lead ALS candidate, Amylyx Pharmaceuticals has picked up $135 million to bankroll a potential commercial launch and support a pipeline of other candidates for neurodegenerative diseases, including the one causing a stir this summer: Alzheimer’s disease.
The Cambridge, Massachusetts-based biotech raked in the oversubscribed series C Tuesday to support the clinical development and launch plans, pending a Canadian green light for the amyotrophic lateral sclerosis (ALS) treatment. The novel combination of compounds called AMX0035 was found to slow the progression of ALS in a phase 2/3 study presented last September.
Viking Global Investors led Tuesday’s financing round, with participation from Bain Capital Life Sciences, Perceptive Advisors and about a dozen other backers.
AMX0035 combines two orphan drugs—taurursodiol and Buphenyl—into an oral medication that works to protect brain cells’ energy-producing mitochondria and bolster the endoplasmic reticulum, ensuring constructed proteins fold and operate correctly.
The therapy was put up for a Health Canada nod last month and Amylyx intends to submit for a marketing approval with the European Medicines Agency by the end of this year. FDA regulatory updates are to come, the company said.
Amylyx also presented a trial design for an international phase 3 study of the drug in May.
The biotech has beefed up its board and leadership in recent months to support a commercial roll-out, including the additions of Amicus Therapeutics CFO Daphne Quimi and Paul Fonteyne, the former president and CEO of Boehringer Ingelheim’s U.S. operations. Amylyx also hired Chris Aiello in April to lead operations in Canada as general manager, after leading the rare disease and rare blood disorder units at Sanofi Genzyme’s Canadian outfit.
Amylyx also landed a new CFO in January with a direct connection to the ALS movement, picking up former Alkermes CFO James Frates, who is the cousin of the driving force behind the Ice Bucket Challenge. Funds raised from the viral ALS awareness social media movement helped support Amylyx’s own trial.
The 2020 Fierce 15 winnerraised $30 million last summer for AMX0035 and a midstage trial for a potential Alzheimer’s treatment. Last November, Amylyx said the last patient had completed the phase 2 trial, dubbed Pegasus, with top-line data due in the first half of this year. The company’s website now says the read out will happen sometime this year.
The company could benefit from the revived interest in Alzheimer’s following the FDA’s controversial approval of Biogen’s Aduhelm. The decision has been criticized by federal, congressional, industry and hospital system stakeholders alike, but competing companies have seen a rise in investment interest.
Amylyx Pharmaceuticals has joined the public markets with a $190 million IPO that will support the company as it shepherds its amyotrophic lateral sclerosis drug through regulatory review, and if all goes well, a commercial launch as a new treatment for the neuromuscular disorder.
Cambridge, Massachusetts-based Amylyx boosted the size of the offering by 1.25 million shares. Initially planning to offer 8.75 million shares, Amylyx ended up offering 10 million shares at $19 apiece, which was the midpoint of the targeted $18 to $20 range. Those shares began trading on the Nasdaq Friday under the stock symbol “AMLX.” There was no pop for Amylyx’s stock price, which closed its first day of trading at $18.07.
Amylyx aims to treat ALS by addressing the death of neurons. The company’s drug, AMX0035, is comprised of two small molecules that each take a different approach to pathways associated with neuronal survival. The company contends that the two mechanisms combined offer the potential to help neurons live longer. In a placebo-controlled Phase 2 study, patients treated with the Amylyx drug showed improvement on measures of physical function in ALS patients. Based on those results, Amylyx submitted a new drug application to the FDA. Last week, the agency accepted that application under priority review and set a June 29 target date for a regulatory decision.
Before the FDA makes a decision, it will convene an independent panel of experts to discuss AMX0035’s safety and efficacy as well as any scientific questions regarding the drug. The date for that advisory committee meeting has not yet been set. In the meantime, a larger Phase 3 study is underway. The FDA went back and forth on whether the larger study was needed to support a drug application. Though the agency ultimately concluded that Amylyx could seek approval based on the Phase 2 data, the company is proceeding with the late-stage study to generate additional data on the drug’s safety and efficacy as it pursues regulatory approvals in other markets around the world.
According to the IPO filing, Amylyx traces its beginnings to a Brown University dorm room where, in 2013, co-founders Josh Cohen and Justin Klee set out to find an answer to the question of why neurons die. The duo’s research led to the development of AMX0035, the company’s only drug candidate so far. Though ALS is the drug’s first disease target, Amylyx notes that improving neuron survival has potential applications in many neurodegenerative disorders. A clinical trial is already underway testing AMX0035 in Alzheimer’s disease and tests in Wolfram disease are also planned.
Since its formation, Amylyx has raised about $234 million, most recently a $135 million Series C round of funding last July. Morningside Venture Investments is Amylyx’s largest stockholder, owning an 18.9% post-IPO stake, according to the prospectus. ALS Invest 1 owns 10.8%, while Viking Global Investors owns 8.8%.
As of the end of the third quarter of 2021, Amylyx reported a $125.7 million cash position. Combined with the IPO proceeds, the company plans to deploy about $100 million of its capital toward the regulatory review process of AMX0035 in ALS, as well as preparation for the potential launch of the drug—if it’s approved. Another $15 million is set aside to fund the ongoing Phase 3 clinical trial through to completion; $10 million is earmarked for expanding the company’s drug pipeline to other neurodegenerative disorders.
After a long road filled with scrutiny and uncertainty, Amylyx’s amyotrophic lateral sclerosis (ALS) drug, known as AMX0035, has finally scored FDA approval.
The drug, now branded as Relyvrio, won approval for the treatment of ALS in adults. It’s the first ALS treatment that showed a significant slowing in both disease progression and functional decline, as well as extended survival, in a randomized clinical trial.
After a first FDA advisory committee meeting in March ended with a negative 6-4 vote for the drug, many companies would have stopped there. But Amylyx was persistent, and the committee reconvened this September to discuss a new analysis the company submitted, resulting in a positive 7-2 vote. The long stretch of time helped the company prepare for an eventual launch, despite not knowing when, or if, its drug would get approval.
“We’ve been planning this a long time,” said Josh Cohen, Amylyx’s co-CEO and co-founder, referring to the drug’s original FDA target decision date of June 29. “We’re fully prepared,” Cohen said in an interview with Fierce Pharma ahead of the approval.
The drug will carry a list price of about $158,000 per year in the U.S., the company said on a post-approval conference call, noting that the price is below the latest FDA-approved ALS product. However, Amylyx is “committed to providing financial assistance” for patients with commercial insurance and will provide the drug at no cost for underinsured or uninsured patients who meet “certain financial eligibility criteria” and who have “exhausted all other options,” said Amylyx’s chief commercial officer Margret Olinger.
The price, before being revealed, was already criticized by U.S. watchdog ICER in June. Using a placeholder price of $169,000 per year, the organization said the cost-effectiveness of the medicine would “far exceed typical thresholds.”
The FDA based its approval on data from the phase 2 Centaur trial and biomarker results from a phase 2 of the drug in Alzheimer’s disease. At the recent FDA expert panel meeting, some panelists voiced a desire to wait for the results of the phase 3 Phoenix trial. The company doesn’t expect top-line results from that trial until 2024, Justin Klee, Cohen’s counterpart as co-CEO and co-founder, said in an interview with Fierce Pharma.
“For people with ALS, that’s a long, long time,” Klee said. If the agency were to wait until the results of that trial came out, “a whole generation of ALS patients will die without access,” Cohen added. “It seems against common sense to deny that to people.”
From pricing to insurance to supply, Amylyx is set and covered, Cohen said. But the company wants to do more than “just launch a drug into the market.”
“ALS has a lot of other things that need changing,” Cohen said. From diagnoses that take too long, “woefully underfunded” clinics, and challenges barring access for patients, there are a number of unaddressed, long-standing problems that need addressing, and that Amylyx wants to “put a pretty significant effort” into fixing.
“ALS has a lot of other things that need changing,” Cohen said. From diagnoses that take too long, “woefully underfunded” clinics, and challenges barring access for patients, there are a number of unaddressed, long-standing problems that need addressing, and that Amylyx wants to “put a pretty significant effort” into fixing.
Amylyx Pharmaceuticals, Inc. (NASDAQ: AMLX) (“Amylyx” or the “Company”) today announced topline results from PHOENIX, a global, 48-week, randomized, placebo-controlled Phase 3 clinical trial of AMX0035 (sodium phenylbutyrate and taurursodiol [also known as ursodoxicoltaurine]; RELYVRIO® in the U.S., ALBRIOZA™ in Canada) in people living with amyotrophic lateral sclerosis (ALS). PHOENIX did not meet its primary endpoint of reaching statistical significance (p=0.667) as measured by change from baseline in the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) total score at Week 48, nor was there statistical significance seen in secondary endpoints. Amylyx plans to present the data from PHOENIX at an upcoming medical meeting and will publish the results in a medical journal later this year.
Amylyx will continue to engage with regulatory authorities and the broader ALS community, including ALS specialists and other multidisciplinary experts, people living with ALS, and advocates, to discuss the results from PHOENIX within the next eight weeks and make informed decisions. Amylyx intends to share plans for RELYVRIO/ALBRIOZA in ALS, which may include voluntarily withdrawing RELYVRIO/ALBRIOZA from the market. At this time, RELYVRIO/ALBRIOZA and its related patient support program will continue to be available for people living with ALS. Amylyx has voluntarily decided to pause promotion of the medication during this time.
We are surprised and deeply disappointed by the PHOENIX results following the positive data from the CENTAUR trial. Our main priority at the moment is sharing the information with people living with ALS and their treating physicians; this is part of our continued commitment to them and our mission. Over the next eight weeks, our team will continue to engage with regulatory authorities and the ALS community to discuss the results from PHOENIX. We will be led in our decisions by two key principles: doing what is right for people living with ALS, informed by regulatory authorities and the ALS community, and by what the science tells us. On behalf of the entire Amylyx team, we are grateful to the ALS community and for the dedication of trial participants, investigators, and study site teams. With data collected from 664 participants in PHOENIX, we are certain there will be important learnings that will help inform future ALS research. We are steadfast in our commitment to the ALS community and our mission, including with AMX0035 where it has shown potential in neurodegenerative diseases such as Wolfram syndrome and progressive supranuclear palsy, and with AMX0114, our investigational antisense oligonucleotide targeting calpain-2, in ALS,” said Justin Klee and Joshua Cohen, Co-CEOs of Amylyx.
PHOENIX Study Results:
The Phase 3 PHOENIX study enrolled 664 adults living with ALS. Participants were randomized three-to-two to receive either AMX0035 or placebo, with both treatment groups receiving standard-of-care. Continuation of a stable dosing regimen of riluzole and/or edaravone was permitted.
PHOENIX did not meet the primary endpoint: There was no significant difference observed between participants treated with AMX0035 and placebo in ALSFRS-R total score change from baseline at Week 48 (p=0.667). No significant difference was observed in the subset of participants who met the CENTAUR trial criteria. There were also no significant differences observed across secondary endpoints.
Consistent safety and tolerability profile: AMX0035 was well-tolerated in PHOENIX. There were no new safety signals, reinforcing the favorable and manageable safety profile observed with AMX0035 to date.
European participants who completed the 48-week randomized phase had the option to enroll in an open label extension of the trial of up to two years in duration, which remains ongoing.
Science of AMX0035:
AMX0035, a specially formulated oral fixed-dose combination of PB and TURSO, has been shown in numerous preclinical studies to have a robust, synergistic effect in targeting two different destructive neurodegenerative disease pathways by mitigating endoplasmic reticulum stress and the associated unfolded protein response and mitochondrial dysfunction thereby reducing neuronal cell death. Additionally, AMX0035 has been shown to also reduce markers associated with neurodegenerative diseases in clinical trials, including a reduction of tau, a key protein aggregate shared across several neurodegenerative diseases, and YKL-40, a marker of neuroinflammation.
Update on Ongoing AMX0035 Studies:
The global, randomized, double-blind, placebo-controlled Phase 3 ORION clinical study of AMX0035 in PSP remains ongoing. The first participant was dosed in December 2023, and the Company is planning for an interim analysis. Topline results continue to be anticipated in 2025 or 2026.
Data from the ongoing 12-participant, single site, open-label Phase 2 HELIOS clinical study are demonstrating evidence of clinical activity of AMX0035 in Wolfram syndrome. This study is fully recruited, and the Company plans to present preliminary data in the second quarter of 2024.
After reporting the failure of a confirmatory trial of its amyotrophic lateral sclerosis (ALS) drug Relyvrio (AMX0035), Amylyx Pharmaceuticals is uncertain whether it will pull the treatment from the market in the U.S. and in Canada, where it is known as Albrioza.
“We’ll spend the next eight weeks engaging with regulatory authorities and the ALS community to share the top-line data,” co-CEO Justin Klee said on a Friday conference call. “We’ll follow the science and do what’s right for the community which may include voluntarily removing the product from the market.”
The 11-year-old Cambridge, Massachusetts-based company has decided to pause promotion of Relyvrio. Patient support services will remain in place, it said.
Two weeks ago, Amylyx reported sales of Relyvrio at $380 million in its first full year on the market, following its approval in September 2022. While sales initially scaled up quickly, the trajectory had slowed, going from $103 million in the third quarter to $108 million in the fourth quarter.
Other than to report that the PHOENIX trial did not meet its primary endpoint, failing to reach statistical significance as measured by change from baseline in the revised ALS functional rating scale (ALSFRS-R), or any of its secondary objectives, Amylyx did not reveal figures from the 48-week trial that enrolled 664 ALS patients.
The company said it will present the data at an upcoming medical conference, and results will be published in a medical journal later this year.
With the devastating news, Amylyx’s shares plummeted 83% by midmorning.
“The news comes as a massive disappointment—not only for us (where AMLX has been a top pick) and investors—but also to ALS patients and the overall ALS community,” analysts from Mizuho Securities wrote in a note to clients.
The company said it will continue to study AMX0035 as a treatment for two other neurodegenerative diseases—Wolfram syndrome and progressive supranuclear palsy. Amylyx also hopes to enter the clinic in the second half of this year with AMX0114, a treatment for ALS patients designed to lower their levels of calpain-2 to strengthen their nerve fibers.
After much consternation and two advisory committee meetings—one voting down AMX0035 6-4 and the other giving it a thumbs-up vote of 7-2 after new analysis of trial data—the FDA signed off on Relyvrio 19 months ago.
The decision was based on results of the phase 2 CENTAUR trial, which showed that AMX0035 slowed progression and functional decline and extended survival. Experts who recommended rejection urged the FDA to wait until results of the phase 3 PHOENIX trial were available.
On Friday, Klee would not speculate on the difference between the trial results.
“I think it’s early,” Klee said. “It’s particularly important to take time to meet with the ALS experts. I think it speaks to the heterogeneity of ALS and also the difficulty in ALS and neurodegenerative diseases more in general. It’s so imperative that when we all, fighting these diseases, have setbacks like this, we learn from them so we can continue to advance because I do very strongly believe that we can have meaningful advances for people with neurodegenerative diseases.”
The ALS Association was heavily invested in the success of the drug as well. Through its Ice Bucket Challenge in 2014, the organization donated $2.2 million to assist in its development. It also collected more than 50,000 signatures and submitted them to the FDA in 2020, urging the regulator for approval.
“While today’s news is disappointing, there are more than 50 potential treatments in the clinical stage of development, including more than a dozen in phase 3 trials,” the ALS Association said in a statement. “We are more committed than ever to ensuring that safe and effective treatments are approved and available to people living with ALS as quickly as possible.”
Merus NV는 Crucell 기술을 기반으로 2003년에 John de Kruif박사와 Ton Logtenberg 가 창업한 회사로서 이 회사의 기술은 Crucell의 PER.C6 cell line을 이용해서 Single Clonal Cell line으로 부터 Oligoclonics를 안정되게 대량으로 생산할 수 있습니다. PER.C6 cell line은 2004년에 Crucell과 DSM으로 부터 기술이전 계약을 받았고 DEKK IgG를 통해 Stable Bispecific Antibody를 만들 수 있는 기반기술을 확보하여 Full-length IgM format으로 Long half-life와 Low immunogenicity라는 중요한 장점을 확보할 수 있었습니다.
Bispecific T-cell engager 기술개발은 생산에 많은 자금이 소요되어 Merus는 매년 $50 Million 이상의 유상 증자나 계약을 통해 자금조달을 하고 있고 Novartis, Johnson & Johnson, Eli Lilly, Ono등의 제약사들이 지분투자나 공동계발에 참여하고 있습니다. 최근에는 Gilead와 $1.5 Billion 공동계약을 한 바 있는데 이를 통해 Merus는 Trispecific T-cell Engager에 영역을 확장하게 되었습니다.
Merus의 Multiclonics Platform은 세가지 기술을 접목하는데 (1) Merus Mouse (MeMo)라는 기술로 다양한 cLC (common Light Chain) antibodies를 mouse 에서 얻고 이들의 타겟을 발굴하고 (2) Robotics를 이용해 cLC antibodies와 DEKK IgG를 결합한 수천개의 Multiclonics를 만들고 unbiased functional screening을 통해 (3) Best-in-class Biclonics나 Triclonics를 발굴하는 것입니다.
현재 자체 개발 중인 프로그램은 Petosemtamab (MCLA-158, EGFR x LGR5), Zenocutuzumab (MCLA-128, HER2 x HER3), MCLA-129 (EGFR x MET)가 임상 1/2에 있고 MCLA-145 (CD37 x PD-L1)이 임상 1상에 있습니다.
공동연구 중인 프로그램은현재 4가지가 있는데, BETTA와 진행 중인 MCLA-120 (EGFR x c-MET)이 임상 1/2상을 진행 중이고 ONO Pharma와 공동개발 중인 ONO-4685 (PD-1 x CD3)가 임상 1상에 Incyte와 개발 중인 INCA32459 (LAG3 x PD-1)과 INCA33890 (TGFBr2 x PD-1)이각각 임상 1상에 진입한 상태입니다.
금년 3월에 발표한 Corporate Presentation에 보다 자세한 설명이 있습니다. Merus NV의 Multiclonics Platform이 이제 Bispecific T-cell Engager 뿐만 아니라 Trispecific T-Cell Engager로 영역을 확장하고 있는데 아직까지는 임상이 초기단계이지만 곧 몇개의 프로그램은 pivotal clinical trials에 도달할 수 있을 것 같아 기대가 됩니다.
Dutch biotechnology company Crucell N.V. (Euronext:CRXL) (Nasdaq:CRXL) and allied contract manufacturer DSM Biologics announced today that Crucell and Merus B.V. have signed a PER.C6(R) research license agreement. This license agreement allows Merus to use the PER.C6(R) cell line for the further development of its Oligoclonics(TM) technology and related products.
“Oligoclonics(TM) are mixtures of human antibodies, produced by a single clonal cell line, that we expect to have improved clinical efficacy compared to current antibody therapeutics,” said Ton Logtenberg, CEO of Merus. “The PER.C6(R) cell line is instrumental in the stable and high yield production of Oligoclonics(TM).”
Under the terms of the agreement, Crucell and DSM will receive an upfront payment and annual maintenance fees. Further financial details were not disclosed.
About Merus
Merus is a Dutch biotechnology company founded in June 2003. The Company is focused on the discovery and development of a novel class of human antibodies, Oligoclonics(TM), that are expected to have improved clinical efficacy compared to current generations of human monoclonal antibodies. For more information, please visit http://www.merus-biopharm.com.
Merus BV, a biopharmaceutical company focused on the discovery and development of mixtures of human therapeutic antibodies, today announced that it has received a grant worth € 0.67 million for the development of an antibody combination therapy for chronic inflammatory diseases using its novel OligoclonicsTM and MeMoTM technologies. The grant was awarded by EuroTransBio.
Merus is applying the OligoclonicsTM and MeMoTM technologies to build a pipeline of innovative human therapeutic antibodies. With these technologies, Merus generates combinations of therapeutic antibodies produced from a single cell that boast superior biological activity when compared to single antibodies. The EuroTransBio grant will allow Merus to develop innovative antibody drugs for the treatment of chronic inflammatory diseases such as Rheumatoid Arthritis.
Ton Logtenberg, CEO of Merus, said: “We are pleased that EuroTransBio has recognized the potential of Merus’s innovative technologies to deliver next generation therapeutics offering substantial clinical benefit to patients with chronic diseases. This grant supports collaboration between European companies and academic institutions that combine their unique expertise to achieve this goal”.
Therapeutic monoclonal antibodies, a highly successful class of biological drugs, are conventionally manufactured in mammalian cell lines. A recent approach to increase the therapeutic effectiveness of monoclonal antibodies has been to combine two or more of them; however this increases the complexity of development and manufacture. To address this issue a method to efficiently express multiple monoclonal antibodies from a single cell has been developed and we describe here the generation of stable cell clones that express high levels of a human monoclonal antibody mixture. PER.C6® cells were transfected with a combination of plasmids containing genes encoding three different antibodies. Clones that express the three corresponding antibody specificities were identified, subcloned, and passaged in the absence of antibiotic selection pressure. At several time points, batch production runs were analyzed for stable growth and IgG production characteristics. The majority (11/12) of subclones analyzed expressed all three antibody specificities in constant ratios with total IgG productivity ranging between 15 and 20 pg/cell/day under suboptimal culture conditions after up to 67 population doublings. The growth and IgG production characteristics of the stable clones reported here resemble those of single monoclonal antibody cell lines from conventional clone generation programs. We conclude that the methodology described here is applicable to the generation of stable PER.C6® clones for industrial scale production of mixtures of antibodies.
Merus NV of Utrecht, the Netherlands, has closed a €21.7 million second financing round led by new investors the Novartis Option Fund, Pfizer Inc., Bay City Capital, and Life Sciences Partners. Merus’ seed investor Aglaia Oncology Fund followed on.
The money will be used to advance development of Merus’ antibody-based drugs for the treatment of cancer, inflammation and infectious diseases.
At the same time, the company granted the Novartis Option Fund the option to an exclusive license to one of Merus’ oncology programmes. The agreement includes upfront and potential milestones payments totalling over S200 million, plus royalties.
“We are very pleased by the high quality of the new investor syndicate,” said Ton Logtenberg, Merus CEO. “The mix of renowned financial and corporate US and European VCs is a validation of the perceived high value of Merus’ technologies.“
The proceeds will enable Merus to demonstrate its bispecific antibodies can target diseases that cannot be treated with single monoclonal antibodies. “The agreement with Novartis Option Fund further underscores that big pharma recognises the potential of our innovative antibody therapeutics,” Logtenberg said.
Lionel Carnot, an investment partner with Bay City Capital, said Merus’ technology, “Is a very ingenious solution to the key issues that have hindered the development of poly- and oligoclonal therapeutics.”
Merus B.V., a biopharmaceutical company focusing on innovative human antibody therapeutics, today announced a €31 million (US$42 million) extension to its Series B financing round, bringing the total round to €47.6 million (US$65 million).
Johnson & Johnson Development Corporation (JJDC) joined as a new investor along with existing investors Novartis Venture Fund, Pfizer Venture Investments, Bay City Capital, LSP (Life Sciences Partners), and Aglaia Oncology Fund. A representative of JJDC will join Merus’ Board of Directors. Merus will use the new funds to broaden its portfolio of pre-clinical programs for the treatment of cancer patients and to bring its lead programs into phase I clinical testing.
“We view the continuing support of our investors as a strong endorsement of our technology, our team and our strategy,” said Ton Logtenberg, Chief Executive Officer of Merus. “We are particularly proud to welcome Johnson & Johnson Development Corporation to our investment consortium as our third big pharma corporate venture investor. As a next milestone, we are looking forward to moving our lead candidate into clinical development next year.”
The investment of JJDC in Merus B.V. was announced at the launch of the Johnson & Johnson Innovation Centre in London today.
Earlier this year, Merus presented encouraging research and preclinical data of MCLA-117, a product candidate to treat acute myeloid leukemia, a disease with very poor long-term prognosis. MCLA-117 is based on Merus’ proprietary Biclonics™ ENGAGE platform and is currently in development.
Merus B.V., a clinical-stage immuno-oncology company developing innovative bispecific antibody therapeutics, today announced that it has entered into an agreement with investors for the sale of up to € 72.8 million ($80.5 million) of Series C preferred shares and consummated the first tranche under the agreement. New investors include Sofinnova Ventures and Novo A/S as the co-leads, along with RA Capital Healthcare Fund, Rock Springs Capital, Tekla Capital Management and an unnamed U.S.-based life sciences-focused investor. The company’s existing investors, including Novartis Venture Fund, Johnson & Johnson Innovation – JJDC, Inc., Pfizer Venture Investments, Bay City Capital, LSP Life Sciences Partners and Aglaia Oncology Fund, also participated in the financing.
“The proceeds from this financing provide us with funding to advance our key clinical and preclinical programs and to broaden our pipeline of innovative therapeutics that recruit cells of the immune system to kill cancer cells,” said Ton Logtenberg, Ph.D., Chief Executive Officer of Merus. “This financing follows the progression of Merus into a clinical stage company. Our first lead bispecific antibody, MCLA-128, has commenced phase 1/2 clinical trials as a potential targeted therapy for solid tumors and our second lead bispecific antibody, MCLA-117 for the treatment of acute myeloid leukemia, is planned to commence clinical trials in the first quarter of 2016.“
As part of the transaction, Anand Mehra of Sofinnova Ventures and Jack Nielsen of Novo A/S have joined the Merus board of directors.
“Cancer remains a disease of significant unmet medical need where targeted therapies that activate the immune system to kill tumor cells hold the promise of providing novel and effective treatment options for patients,” said Anand Mehra. “Merus’ proprietary technology platform has enabled the company to build a significant pipeline of promising immuno-oncology drug candidates.”
About Merus B.V. Merus is a clinical-stage immuno-oncology company developing innovative bispecific antibody therapeutics, referred to as Biclonics. Biclonics are based on the full-length IgG format, are manufactured using industry standard processes and have been observed in preclinical studies to have several of the same features of conventional IgG-based antibodies, such as long half-life and low immunogenicity. Merus’s lead Biclonics product candidate, MCLA-128 is being evaluated in a Phase 1/2 clinical trial in Europe as a potential treatment for HER2-expressing solid tumors. Merus’s second Biclonics product candidate, MCLA-117, is being developed as a potential treatment for acute myeloid leukemia, and Merus expects to initiate clinical trials of this candidate in the first quarter of 2016. The company also has a robust pipeline of proprietary product candidates in pre-clinical development, including Biclonics designed to bind to various combinations of immunomodulatory molecules, including PD-1 and PD-L1. For further information, please visit www.merus.nl.
Merus is edging toward its long-gestating $65 million (€57 million) Nasdaq IPO. The cancer specialist, which lists Novartis ($NVS), Johnson & Johnson ($JNJ) and Pfizer ($PFE) among its largest investors, plans to build the IPO on the support of its existing backers, which are expected to buy half of the offered shares.
Utrecht, the Netherlands-based Merus first talked up the prospect of a Nasdaq IPO during the go-go month of April 2015. At that time, the $100 million it was aiming for was on the low side of the sums being proposed and raised by fellow oncology biotechs such as Adaptimmune Therapeutics ($ADAP), Aduro Biotech ($ADRO) and Blueprint Medicines ($BPMC). Yet by the time Merus formalized its plan to go public in October, the tide had turned, leaving a clutch of biotechs on the outside looking in through the then-closed IPO window.
Having raised €72.8 million in a private round in August, Merus was better equipped than some to ride out the downturn in sentiment toward biotech IPOs. And the immuno-oncology player has gone back to its current investors to get its IPO out the door. Merus has commitments from as-yet-unnamed existing institutional investors to buy $32.5 million of the IPO shares. Bay City Capital, Aglaia Oncology Fund, Sofinnova Venture Partners, Novo A/S and LSP sit alongside the aforementioned Big Pharma trio on the list of Merus’ main investors. Each organization owns more than 5% of the company.
If Merus can find buyers for the remaining 2.2 million shares at the $15-a-pop midpoint of its target range, it will exit the offering with net proceeds of $56.7 million, a sum it plans to split fairly evenly between three of its pipeline prospects. Lead candidate MCLA-128 will swallow up $17 million of the IPO haul. Merus anticipates this funding will take it through to the end of a Phase I/II clinical trial in patients with HER2-expressing solid tumors. Merus expects top-line data in the second half of 2017, but that target that has yo-yoed over the past 6 months.
As recently as January, Merus told investors to expect data by the end of the year. While the public is now set to have to wait longer to get a full look at the data, Merus is continuing to release snapshots of the progress of the study. In the dose-escalation stage, 12 of the 27 patients who took MCLA-128 experienced “an objective positive response.” Of those 12, 11 had stable disease after two cycles of treatment. Three people were stable after four rounds of therapy. Merus thinks MCLA-128 kills cancer cells by blocking growth pathways and eliciting the support of immune effector cells.
Merus is moving another two candidates down the pipeline closely behind MCLA-128. The more advanced of the pair, MCLA-117, began a Phase I/II study in patients with acute myeloid leukemia this month. If Merus hits its IPO fundraising target, it will commit $14 million to the study, a sum it thinks will see it through to the delivery of top-line data in the first half of 2018. A further $10 million is earmarked for MCLA-158, which is expected to enter the clinic as a treatment for colorectal cancer before the end of 2017. Each of the candidates is a bispecific antibody.
The willingness–or not–of Nasdaq investors to bankroll the advance of the programs could go some way to indicating whether the freeze on listings by European biotechs is coming to an end. Over the past 6 months, Basilea Pharmaceutica (SWX:BSLN) and Bavarian Nordic (CPH:BAVA) have both canned planned listings, while perennial IPO bridesmaid Mapi Pharma has continued to try unsuccessfully to go public.
Incyte Corporation (NASDAQ:INCY) and Merus N.V. (NASDAQ:MRUS) announced today that they have entered into a global, strategic collaboration agreement focused on the research, discovery and development of bispecific antibodies utilizing Merus’ proprietary Biclonics® technology platform. The Collaboration and License Agreement grants Incyte the exclusive rights for up to eleven bispecific antibody research programs, including two of Merus’ current preclinical immuno-oncology discovery programs.
Biclonics® retain the IgG format of antibodies that are produced naturally by the immune system and, by binding to two targets, enable multiple modes of action that cannot otherwise be obtained with conventional monoclonal antibodies.
“By virtue of a unique ability to simultaneously engage multiple protein targets, we believe bispecific antibodies have the potential to play an important role in the future of biotherapeutics,” said Reid Huber, Ph.D., Incyte’s Chief Scientific Officer. “This collaboration with Merus expands our large molecule discovery capabilities into an innovation-rich area of research, creating additional opportunities for us to deliver on our commitment to improving and extending the lives of patients with cancer and other serious diseases.”
“This transformative, global collaboration further underscores the potential of Merus’ Biclonics® technology platform and establishes a strong relationship with Incyte, a leader in innovative drug development,” said Ton Logtenberg, Ph.D., Chief Executive Officer of Merus. “We look forward to expanding our pipeline under this agreement, as we efficiently exploit our preclinical discovery engine and progress our most advanced, proprietary assets in the clinic.”
Terms of the Collaboration
Under the terms of the collaboration, Incyte has agreed to pay Merus an upfront payment of $120 million. In addition, Incyte has agreed to purchase 3.2 million shares of Merus stock at $25 per share, for a total equity investment of $80 million.
The parties have agreed to collaborate on the development and commercialization of up to 11 bispecific antibody programs. For one current preclinical program, Merus will retain all rights to develop and commercialize approved products in the United States, and Incyte will develop and commercialize approved products arising from the program outside the United States. Following any regulatory approval of a product candidate for this particular pre-clinical program, each company has agreed to pay the other tiered royalties ranging from 6 to 10 percent on net sales of products in their respective territories.
Merus also has the option to co-fund development of product candidates arising from two other programs. For any program for which Merus exercises its co-development option, Merus would be responsible for 35 percent of global development costs in exchange for a 50 percent share of U.S. profits and losses and tiered royalties ranging from 6 to 10 percent on ex-U.S. sales by Incyte for these programs. Merus also has the right to elect to provide up to 50 percent of detailing activities for product candidates arising from one of these programs in the United States.
For each of the other eight programs, Incyte has agreed to independently fund all development and commercialization activities. For these programs, Merus will be eligible to receive potential development, regulatory and sales milestone payments of up to $350 million per program, which could result in an aggregate milestone opportunity of approximately $2.8 billion if all development, regulatory and sales milestones are achieved across all such eight other programs in all territories. Merus will also be eligible to receive tiered royalties ranging from 6 to 10 percent on global sales of any approved products under these eight programs.
Merus will retain rights to both of its clinical candidates and MCLA-158, as well as its technology platform and future programs emerging from Merus’ platform that are outside the scope of this agreement.
The transaction is expected to close in the first quarter of 2017, subject to the early termination or expiration of any applicable waiting periods under the Hart-Scott Rodino Act and customary closing conditions.
Merus N.V. (Nasdaq: MRUS), a clinical-stage bispecific antibody company developing Biclonics®, today announced the pricing of an underwritten public offering of 4,750,000 common shares, at a public offering price of $14.50 per share, before underwriting discounts and commissions. Merus also granted the underwriters a 30-day option to purchase up to an additional 712,500 common shares. The gross proceeds from the offering, before deducting underwriting discounts and commissions and estimated offering expenses, are expected to be approximately $68.9 million, excluding any exercise of the underwriters’ option to purchase additional common shares. All of the shares in the offering are to be sold by Merus.
Eli Lilly, via its Loxo Oncology biotech unit, is signing up to a three-therapy deal with Merus focused on T-cell redirecting bispecific antibody work.
Netherlands-based Merus gets $40 million upfront and a $20 million equity investment from the Big Pharma as well as $1.6 billion in total for three drugs.
These will come out of Merus’ so-called Biclonics platform, which develops CD3-engaging, T-cell redirecting bispecific antibody therapies.
“CD3-engaging bispecific antibodies are rapidly becoming one of the most transformative immune-modulating modalities used to treat cancer,” said Jacob Van Naarden, M.D., chief operating officer of Loxo Oncology.
“We expect these therapies will become an important component of the Loxo Oncology at Lilly biologics strategy. Merus has built a differentiated platform and one that we believe can enable us to create bispecific antibody therapies with wider therapeutic indexes than those available today. We look forward to working closely with Merus to develop new potential medicines for patients with cancer.”
Merus attracted some buzz a few years back with its impressive list of big-name backers including Novartis, Johnson & Johnson and Pfizer.
It had to dial down its plan to raise $100 million in an initial public offering but still managed to pull in $55 million in May 2016. Later that year, it inked a $200 million deal with Incyte to develop bispecific antibodies. Lilly becomes the latest to team up with its platform.
Merus is working on its own internal pipeline focused on zenocutuzumab (also called MCLA-128), targeting fusions involving the gene NRG1, which can drive the growth of many different types of cancers.
“The collaboration with Loxo Oncology at Lilly and their world class research capabilities opens up exciting possibilities for Merus’ Biclonics platform,” added Bill Lundberg, M.D., president and CEO of Merus.
“Our CD3 T-cell engager platform includes over 175 novel and diverse anti-CD3 common light chain antibodies across a wide range of affinities and attributes and enables functional screening of large libraries for optimal performance. We look forward to working together with Loxo Oncology at Lilly to define a new generation of medicines to treat cancer.”
Merus N.V. (Nasdaq: MRUS) (“Merus”, “we” and “our”), a clinical-stage oncology company developing innovative, full-length multispecific antibodies (Biclonics® and Triclonics™), today announced the pricing of an underwritten public offering of 4,848,485 common shares at a public offering price of $24.75 per share. The gross proceeds from the offering, before deducting underwriting discounts and commissions and estimated offering expenses, are expected to be approximately $120 million. In addition, Merus granted the underwriters a 30-day option to purchase up to an additional 727,272 common shares at the public offering price, less the underwriting discounts and commissions. All of the shares in the offering are to be sold by Merus.
Merus N.V. (Nasdaq: MRUS) (“Merus”, the “Company,” “we” and “our”), a clinical-stage oncology company developing innovative, full-length multispecific antibodies (Biclonics® and Triclonics™), today announced the pricing of an underwritten public offering of 3,859,650 common shares, at a public offering price of $28.50 per share (the “Offer Shares”). Merus also granted the underwriters a 30-day option to purchase up to an additional 578,947 common shares (the “Option Shares” and together with the Offer Shares, the “Shares”). The gross proceeds from the offering, before deducting underwriting discounts and commissions and estimated offering expenses and excluding the underwriters’ option to purchase the Option Shares, are approximately $110.0 million. All of the shares in the offering are to be sold by Merus.
Merus N.V. (Nasdaq: MRUS) (“Merus”, the “Company,” “we” and “our”), a clinical-stage oncology company developing innovative, full-length multispecific antibodies (Biclonics® and Triclonics®), today announced the pricing of an underwritten public offering of 6,818,182 common shares, at a public offering price of $22.00 per share (the “Offer Shares”). Merus also granted the underwriters a 30-day option to purchase up to an additional 1,022,727 common shares (the “Option Shares” and together with the Offer Shares, the “Shares”). The gross proceeds from the offering, before deducting underwriting discounts and commissions and estimated offering expenses and excluding the underwriters’ option to purchase the Option Shares, are expected to be approximately $150.0 million. All of the shares in the offering are to be sold by Merus.
Gilead Sciences is partnering with Netherlands-based clinical-stage oncology biotech Merus to find new dual tumor-associated antigens targeting tri-specific antibodies, the companies announced Wednesday.
Under the deal, Merus will receive an upfront payment of $56 million for initial targets as well as an equity investment from Gilead of $25 million in Merus common shares. Merus has the potential to receive up to $1.5 billion in additional payments based on potential development and commercialization milestones.
The research collaboration, option, and licensing agreement will leverage Merus’s proprietary platform. According to the company, the platform can design antibodies capable of binding to three targets simultaneously.
“We have seen the successful application of bispecific antibodies as an immune-modulating modality used to treat cancer. We are now looking ahead to developing additional multispecific antibodies capable of driving robust anti-tumor immune responses with an improved efficacy and safety profile,” Flavius Martin, executive vice president of Research at Gilead, said in a statement. “We are excited to explore the potential of Merus’ differentiated Triclonics platform to discover and advance transformative new cancer therapies as we deepen our portfolio across oncology indications.”
The move comes as Gilead has been on a dealmaking path in 2024. In February, the Bay Area-based biotech announced a $4.3 billion acquisition of CymaBay Therapeutics, snapping up its lead candidate seladelpar which designed to treat the autoimmune disease primary biliary cholangitis.
“We are looking forward to working with Gilead to develop novel T-cell engager antibodies using our Triclonics technology,” Merus Chief Business Officer Hui Liu said in a statement. “We are grateful for our collaborations which represent opportunities for Merus to leverage our research capabilities to pursue innovative biology and to address significant unmet medical needs. Importantly, this collaboration represents the first for our proprietary Triclonics platform.”
The collaboration will see some competition as it looks to target the T-cell engager space. Amgen announced in October 2023 that its bispecific T-cell engager showed positive Phase II results. The tarlatamab drug was investigated in patients with small cell lung cancer with advanced disease and, had an objective response rate of 40% and hit the primary endpoint. Median progression-free survival was under five months while the median overall survival was 14.3 months.
In related news, Crossbow Therapeutics announced Tuesday that it has nominated its first development candidate, a T-cell engager.
Arvinas는 Yale University Craig M. Crews 교수 연구실에서 개발한 PROTAC을 상용화 하기 위해 2013년에 New Haven, Connecticut에서 설립한 회사로 지금까지 10년간 Merck, Roche/Genentech, Pfizer, Bayer 등과 공동연구계약 및 VC/Crossover Funding을 통해서 성장한 회사이다.
PROTAC은 2001년에 PNAS 논문에 처음으로 보고한 이래 2024년 현재 Small molecule 분야 중에서 가장 핫한 분야 중 하나이고 Arvinas가 가장 앞선 회사로 평가한다.
Pfizer와 공동개발 중인 Vepdegestrant (ARV-471)가 현재 ER+/HER2- Breast cancer 치료제로서 임상3상이 진행 중이다.
Arvinas는 2013년에 창업한 이래 PROTAC 분야의 역사를 쓰고 있는 중이다. Arvinas의 신약개발이 잘 순항해서 Oncology 분야 뿐 아니라 Neurology 분야 등에서도 환자들의 치료에 쓰일 수 있기를 기대한다.
Arvinas Inc., a biotechnology company creating a new class of drugs based on protein degradation, today announced it has raised $15 million in Series A funds and $4.25 million in financial support, $1 million of which is in the form of equity, from the Connecticut Department of Economic and Community Development and Connecticut Innovations. Investors in the Series A round include co-leads Canaan Partners and 5AMVentures along with Connecticut Innovations and Elm Street Ventures. The funds will support the development of the company’s technology which has primary application in multiple oncology indications and potential in inflammatory, autoimmune and rare diseases.
Arvinas is built on the research of Craig Crews, PhD, Lewis B. Cullman Professor of Molecular, Cellular and Developmental Biology and professor of Chemistry and Pharmacology at Yale University. The new drugs being developed by Arvinas would induce a cell’s own protein-degradation capabilities to bind to a particular protein and “label” it for degradation, thus removing a protein from the system entirely. This contrasts to a more traditional drug development approach that inhibits proteins. However, only 25 percent of the body’s 20,000 proteins can be inhibited. Proteins that cannot be inhibited can potentially be degraded using Arvinas’ approach, radically expanding the number of disease-causing proteins that can become the targets of new drugs.
“Degrading proteins as opposed to inhibiting them has potential to open up areas of drug development that were previously closed because of the technical limitations of protein inhibition,” said Tim Shannon, MD, CEO of Arvinas and Venture Partner at Canaan Partners. “The Arvinas technology platform represents an entirely new class of drugs bringing an innovative approach to treating disease.“
“In addition to the fact that a very large portion of proteins cannot be blocked, inhibition is not permanent, so a disease-causing protein can eventually become active again after treatment with a drug,” said Dr. Crews. “To effectively stop cancer, a drug-binding site must be inhibited 95 percent of the time, which is currently difficult to achieve. If a protein is removed entirely, that should overcome this problem.”
Arvinas also announced the formation of a Scientific Advisory Board (SAB), which will help guide the development of its novel approach. Members of the SAB include Daniel D. Von Hoff, MD, Chief, distinguished professor and director of clinical translational research division at the Translational Genomics Research Institute and Chief Scientific Officer for US Oncology; Mark Murcko, PhD, former Chief Technology Officer at Vertex Pharmaceuticals; Thomas J. Lynch, Jr., MD, Director of the Yale Cancer Center and Physician-in-chief at Smilow Cancer Hospital at Yale-New Haven; Richard Ulevitch, Venture Partner 5AM and Professor and Chairman Emeritus of the Department of Immunology at The Scripps Research Institute, La Jolla, California; and Peter Farina, PhD, executive in residence at Canaan Partners and former Senior Vice-President of Development at Boehringer Ingelheim.
Arvinas worked with the Yale Office of Cooperative Research (OCR) to secure intellectual property protection for the technology.
“The Arvinas team has lined up an impressive slate of supporters of the unique technology that comes out of Yale University,” noted John Soderstrom, PhD, Managing Director of the Office of Cooperative Research at Yale and a member of Arvinas’ Board of Directors. “Degrading proteins that are driving disease has the potential to bring about drastic changes in drug development, and we anticipate significant interest from pharmaceutical companies.”
Joining Dr. Shannon and Dr. Soderstrom on the Arvinas Board of Directors will be Kush Parmar, MD PhD and a Principal at 5AM Ventures and Brad Margus, the CEO of Genome Bridge and former CEO of Envoy Therapeutics.
Under the deal, Merck will hand the New Haven, CT, biotech an up-front payment and research funding, promising more cash tied to development milestones and setting Arvinas’ maximum haul at $434 million if everything works out over the multiyear agreement. In exchange, Merck will get a chance to use the company’s proteolysis-targeting chimera, or PROTAC, technology, which creates small-molecule treatments that mark proteins for degradation.
Based on work out of Craig Crews’ Yale University labs, PROTAC treatments are designed to get rid of unwanted proteins by triggering a cell’s natural clean-up system, marking targets for removal and letting the body’s degradation mechanisms do the rest. Arvinas, launched in 2013, has largely focused its internal efforts on oncology, but the Merck deal spans multiple disease targets in an undisclosed array of therapeutic areas, the company said.
The majority of protein-targeting therapeutics in the market or in development work by either inhibiting or boosting their targets, whether via antibodies or small-molecule chemicals. But only about a quarter of the body’s roughly 20,000 proteins can be effectively drugged that way, Arvinas CEO Manny Litchman said. By attacking proteins from within their home cells, however, Arvinas’ technology can potentially open up new avenues of therapeutic development, he said, exposing some long-untouchable targets to guided degradation.
That potential was a major selling point for Merck, Litchman said, and now it’s on Arvinas to demonstrate that its technology can come through in the proof-of-concept stage, rolling into what the CEO expects to be “a true collaboration.” Merck has the option to expand the deal to include more disease targets, triggering an undisclosed payment, and Litchman believes the agreement could create a model for Arvinas’ future partnerships.
The company has held onto a host of internal programs also based on PROTAC, including a lead oncology asset Arvinas expects to get into the clinic in the middle of next year. The biotech will likely look to ink one or two more deals along the way, Litchman said, at once cautious not to spread itself too thin and optimistic that Merck’s big co-sign will help it bring would-be partners to the table.
“I think when a company like Merck has done deep due diligence, surveyed the competitive landscape and selected Arvinas as the best platform out there for protein degradation, that’s a signal for others we’ve talked to that perhaps a deeper dive may be warranted,” Litchman said.
Arvinas got rolling with a $15 million A round from Canaan Partners and 5AM Ventures, licensing Crews’ technology and assembling a team of investigators to push it forward. The biotech quickly moved to establish preclinical proof of concept for PROTAC, recruiting Litchman, an 18-year Novartis ($NVS) veteran, in time to start showing off the platform at January’s JP Morgan Healthcare conference.
Fresh on the heels of its inclusion in this year’s Fierce 15, New Haven, CT-based Arvinas has pulled the wraps off a new partnership with Genentech that comes with a $300 million package of milestones.
Genentech, a marquee player in the cancer drug R&D arena, is turning to Arvinas for protein degradation platform tech that was originally developed by Yale’s Craig Crews. Arvinas was launched in 2013 and later signed a development pact with Merck.
The biotech has been focused on moving beyond protein inhibition–a big field in biotech–and into protein degradation, targeting particular proteins for destruction in search of a more permanent solution to a wide array of disease triggers.
“There’s huge interest in this area,” company Chairman Tim Shannon told FierceBiotech earlier in the week. “We’ve had a lot of outreach and we expect more.”
There’s no news on exactly what Genentech is targeting initially, but the biotech arm of Roche has rights to expand the collaboration to include more targets. The upfront in the deal was not disclosed.
“Genentech is very interested in protein degradation as a therapeutic approach to address difficult disease targets,” noted Genentech’s chief deal maker, James Sabry. “Arvinas’s PROTAC technology offers an exciting opportunity to harness the body’s own system to degrade pathogenic proteins.“
The company has been funded by Canaan Partners, 5AM Ventures, Connecticut Innovations and Elm Street Ventures.
Arvinas LLC (“Arvinas”), a private biotechnology company creating a new class of drugs based on targeted protein degradation, today announced that it has closed a Series B financing round of $41.6 million.
All of the initial Series A investors, including the two lead Series A investors, Canaan Partners and 5AM Ventures, participated in this new round. Three additional leading private venture investment firms joined the round: RA Capital Management, OrbiMed, and New Leaf Venture Partners.
“We were impressed by the scientific accomplishments of Arvinas in their first two years and enthusiastic about the robust pipeline entering clinical trials in 2016,” said Andrew Levin, M.D., Ph.D., of RA Capital Management, which led the Series B financing. “Arvinas has a truly unique platform degrading targets of interest, within and outside of oncology, and they are using this powerful platform to rapidly build a portfolio of bifunctional small molecules. We are pleased to join them in this endeavor.”
Arvinas is harnessing the body’s own natural degradation and removal system to target and degrade pathogenic proteins by using bifunctional small molecules, Proteolysis-Targeting Chimeras (PROTACs). PROTACs recruit an E3 ubiquitin ligase to a specific targeted protein, labeling that protein for elimination by the ubiquitin/proteasome system.
In addition to financial resources, this round of investment brings impressive experience and intellectual resources to Arvinas in the form of three new members of the Board of Directors:
Andrew Levin, M.D., Ph.D., Managing Director, RA Capital Management
Stephen Squinto, Ph.D., Venture Partner, OrbiMed
Liam Ratcliffe, M.D., Ph.D., Managing Director, New Leaf Venture Partners
Manuel Litchman, M.D., President and Chief Executive Officer of Arvinas commented: “We are gratified by the continued support of our Series A investors and thrilled with our new investment partners. Andrew, Steve, and Liam bring remarkable track records of accomplishment and knowledge to our Board; they, along with the resources of their firms, will help us succeed as we move forward. The Series B gives us the capital we need to advance an aggressive pipeline of targeted degraders into the clinic and to continue to strengthen our unique platform.”
“This has been a great month for Arvinas, announcing a collaboration with Genentech, being named a ‘Fierce 15’ biotech, and now completing an investment round with several marquee firms,” said Tim Shannon, M.D., Chairman of the Board of Arvinas and General Partner, Canaan Partners.
Two months after expanding a licensing deal with Genentech, privately-held Arvinas LLC struck another lucrative deal with a major pharmaceutical company. Connecticut-based Arvinas inked a deal with Pfizer worth up to $830 million to develop small molecules that can degrade proteins.
Pfizer will use Arvinas’ proprietary PROTAC (PROteolysis TArgeting Chimeras) Platform to create small molecule therapeutics aimed at degrading disease-causing cellular proteins. The two companies provided some brief outlines of the deal but much of the meat was left undisclosed. For example, the companies did not disclose what targets the therapy would take aim at, nor did they disclose how many targets are included in the deal. What is known is that Arvinas will drive discovery efforts and Pfizer will be accountable for the therapy when it reaches the clinical stage and any potential commercialization.
John Ludwig, head of medicinal sciences at Pfizer, said the company has “considerable interest” in protein degradation. He did say the global pharma company would determine the applicability of Arvinas’ PROTAC Platform across multiple therapeutic areas, but did not name them.
Unlike inhibitors, Arvinas’ PROTAC Platform is designed to remove target proteins. The company believes this offers several advantages over traditional small cell inhibitors. By removing target proteins directly rather than simply inhibiting them, PROTACs can provide multiple advantages over small molecule inhibitors which can require high systemic exposure to achieve sufficient inhibition, often resulting in toxic side effects and eventual drug resistance, according to Arvinas data. With multiple protein targets, Arvinas’ PROTAC platform has demonstrated that a transient binding event at a range of binding sites and affinities can translate into very potent degradation of the target protein, the company said. The platform was developed in the Yale University laboratory of Craig Crews, who is the company’s founder and chief scientific advisor.
“As a global industry leader, Pfizer is uniquely positioned to partner with us as we exploit the potential of PROTACs in multiple disease areas,” Arvinas Chief Executive Officer John Houston said in a statement.
Under terms of the deal, Arvinas could receive up to $830 million when all payments, including upfront monies and milestones are factored into the equation. However how those payments will be broken down were not disclosed. If any of the therapies make it to commercialization, Arvinas may be entitled to receive tiered royalties based off any sales.
“This marks another key milestone as we continue to expand the use of our targeted protein degradation platform and advance Arvinas’ first candidates into the clinic.”
Like Pfizer, Genentech has also been tight-lipped about its collaboration with Arvinas. The Bay Area company has not disclosed disease targets it is working on with Arvinas.
In its own pipeline development, Arvinas is focused on targeting both prostate and breast cancer with a focus on androgen and estrogen receptor degradation. In November, the company named its first clinical candidate ARV-110, designed to target and induce degradation of the androgen receptor protein. In December, the company announced its second candidate for clinical development, ARV-378. The candidate is an orally bioavailable small molecule PROTAC designed to target and induce the degradation of the estrogen receptor (ER) protein, which plays a prominent role in the development of ER positive breast cancer.
Former Fierce 15 winner Arvinas, which has caught the attention of Pfizer and Roche over the last year, has got off a strong $55 million series C as it looks to bring its cancer candidates into the clinic.
The New Haven, Conn.-based biotech is, like a number of startups, working on protein degradation, with early-stage efforts focused on oral programs in castration-resistant prostate cancer and the estrogen receptor for ER-positive positive breast cancer.
Both are preclinical, but with this cash boost the biotech is plotting clinical studies in the fourth quarter.
The $55 million round was led by new investor Nextech Invest, with help from Deerfield Management, Hillhouse Capital and Sirona Capital, as well as original investors Canaan Partners, 5AM Ventures, RA Capital Management, OrbiMed and New Leaf Venture Partners.
The cash will also be used to “advance the company’s early-stage oncology pipeline, CNS pipeline and efforts on undruggable targets,” according to a statement.
This comes after a good 12 months for the biotech: In January, ahead of the J.P. Morgan biotech event, Arvinas penned a deal potentially worth $830 million, and more besides, with Big Pharma Pfizer in a pact that centers on the discovery and development of PROTACs (proteolysis targeting chimeras) across multiple disease areas.
And a few months before, in November last year, it inked a deal with Genentech, which saw Roche’s biologics arm double the size of its original alliance with Arvinas, moving the potential value of the pact up above $650 million.
The expansion of the deal allows Genentech to use Arvinas’ protein degradation technology against additional disease targets, also using PROTACs.
This comes as protein degradation is becoming a bigger deal among several smaller biotechs. Fellow Fierce 15 company C4 Therapeutics is tackling protein degradation using small-molecule binders, dubbed degronimids, that can target, destroy and clear proteins through the ubiquitin/proteasome system.
“This past year has been exciting for us with two clinical candidate nominations, the expansion of our collaboration with Genentech and the announcement of a new collaboration with Pfizer,” said John Houston, Ph.D., president and CEO of Arvinas, in the announcement.
“With this additional financial support from existing and new investors who believe in our innovative protein degradation platform, we will continue executing on our strategy of progressing our lead programs to the clinic, expanding the use of the platform outside of oncology, and tackling undruggable targets,” Houston said.
Arvinas has been on a roll this year, starting with a Pfizer R&D deal worth potentially $830 million and bagging a $55 million series C in April to push its cancer drugs into the clinic. Now, the Yale spinout has filed to raise up to $100 million in its IPO, which will get its lead assets through the IND stage and into phase 1.
The New Haven, Connecticut-based biotech is working on an androgen receptor program, ARV-110, in castration-resistant prostate cancer, and an estrogen receptor program, ARV-471, in metastatic ER-positive breast cancer. The bulk of the IPO funds is earmarked to carry these assets into the clinic; what’s left will go toward expanding its protein degradation platform and conducting preclinical work for its earlier-stage programs, Arvinas said in its S-1, filed Thursday.
Arvinas drew its series C round from the likes of Deerfield, Sirona Capital, Canaan Partners and OrbiMed, saying at the time that it aimed to start clinical studies in the fourth quarter.
Arvinas’ drugs are based on its PROTACs (proteolysis targeting chimeras) platform, which grew out of the work of Craig Crews’ lab at Yale University. PROTACs work by activating the body’s protein disposal system. They recruit an enzyme to tag target proteins for ubiquitination and degradation.
Ubiquitination is a process whereby a damaged or unneeded protein is tagged with the protein ubiquitin and then sent to a protein complex called a proteasome, where it is degraded. The hope is that by degrading proteins instead of just blocking them, Arvinas’ drugs will surmount challenges that come with small-molecule protein inhibitors.
The company’s pipeline focuses on cancer, but its multiyear deal with Pfizer centers on developing PROTACs for multiple disease areas. The pair didn’t disclose which indications they would be chasing, but protein degradation could have applications in central nervous system disorders and rare diseases.
Arvinas isn’t alone in the growing protein degradation field. Its Fierce 15 peer, C4 Therapeutics, is using small-molecule binders called degronimids that can target, destroy and clear proteins through the ubiquitin/proteasome system. And Cedilla Therapeutics, which launched in April, is studying protein stability in search of points in the protein degradation process it can intervene.
“Where we are looking is in pivotal events upstream of this machinery that govern the transition [of proteins] between an operational state to a susceptible state,” said Cedilla’s Chief Scientific Officer Brian Jones at the company’s launch.
Bayer and Arvinas, Inc. (Nasdaq: ARVN), a biopharmaceutical company creating a new class of therapies to degrade disease-causing proteins, today announced an agreement to leverage Arvinas’ novel PROTAC® protein degrader technology to develop new human therapeutics for patients with cardiovascular, oncological, and gynecological diseases. In addition, Bayer and Arvinas will jointly launch a new company to leverage Arvinas’ PROTAC® technology for agricultural applications. The overall series of arrangements includes over $110 million in upfront cash and committed funding for the human disease collaboration, the agricultural joint venture, and a direct equity investment by Bayer in Arvinas.
The multi-faceted deal will extend the application of targeted protein degradation to new therapeutic areas and outside human biology. It leverages Arvinas’ expertise in targeted protein degradation, a field the company has led since its founding in 2013, and Bayer’s decades of experience in developing both human therapies and innovative, sustainable agricultural technologies.
“As the first company founded around targeted protein degradation, we’ve been excited about the potential to improve the lives of patients since our inception,” said John Houston, Ph.D., President and Chief Executive Officer of Arvinas. “However, we’ve known that the potential of this technology could be broader than drug development. Through these transactions, not only do we plan to expand our reach into new therapeutic areas, but we and Bayer expect to be the first to apply this approach to agriculture, working to safely and efficiently feed the world’s growing population. It’s a natural next step in our commitment to improving human health.”
“With our unique position as a leading company in both Crop Science and Pharmaceuticals, we see a great opportunity to partner with the pioneer of the PROTAC® technology, to advance this technology as quickly as possible to deliver future solutions for sustainable agriculture and innovative medicines for patients,” said Kemal Malik, Bayer Board member for Innovation.
Pharmaceutical Collaboration and Equity Investment Bayer and Arvinas will collaborate to seek to develop a series of novel product candidates for diseases with serious unmet need. Arvinas will receive an upfront payment and committed R&D funding, as well as a direct equity investment in Arvinas. Combined, these committed funds exceed $60 million. Bayer will own the rights to novel lead structures generated in the collaboration. As programs progress through research, development, and commercialization, Arvinas is also eligible to receive development milestones of over $685 million and commercial royalties ranging from the mid-single digits to the low double-digits.
Agricultural Joint Venture In launching a joint venture (JV), Bayer and Arvinas are investing in one of the greatest challenges facing the world: feeding the growing global population. PROTAC® targeted protein degraders have the potential to address resistance mechanisms in plants to existing agricultural solutions, with solutions to control weeds, insects, and disease by leveraging the selectivity and other features of PROTAC® protein degraders. The JV will be committed to leveraging Arvinas’ PROTAC® protein degrader technology to create innovative, safe, and sustainable agricultural products. The JV will be supported by intellectual property and over $55 million in committed funding from Bayer, and by technology and intellectual property from Arvinas. Bayer and Arvinas will equally share governance and equity ownership of the JV.
Arvinas, Inc. (Nasdaq: ARVN), a biotechnology company creating a new class of drugs based on targeted protein degradation, today announced the pricing of an underwritten public offering of 4,545,455 shares of its common stock at a price of $22.00 per share, before underwriting discounts and commissions. In addition, Arvinas has granted the underwriters an option for a period of 30 days to purchase up to an additional 681,818 shares of common stock at the public offering price, less the underwriting discounts and commissions. All of the shares are being offered by Arvinas.
 Curocell: Anbalcabtagene autoleucel is a novel anti-CD-19 CAR-T therapy – 큐로셀 안발셀")

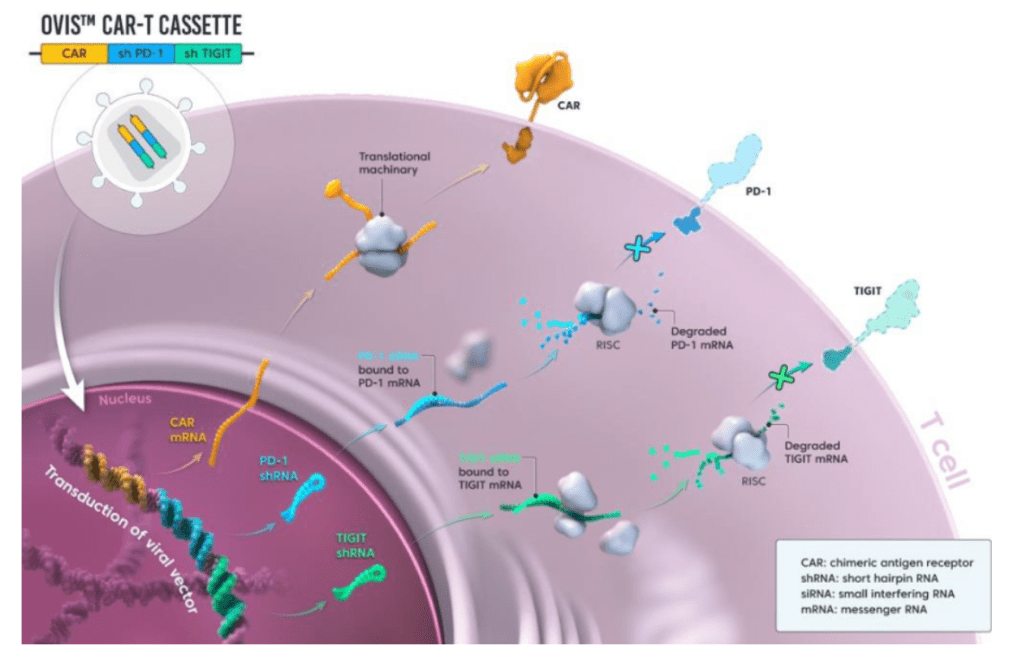



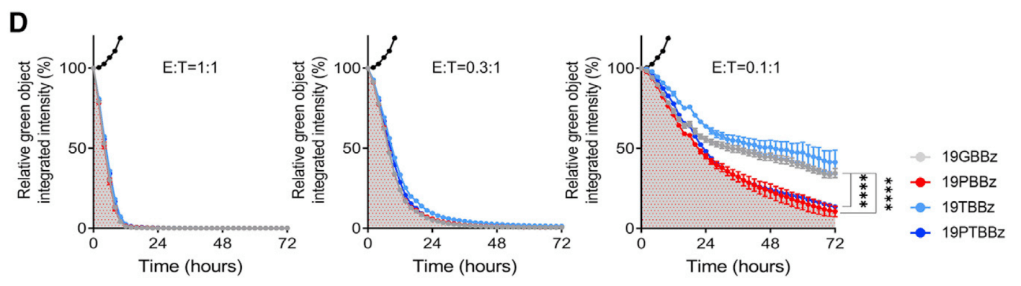
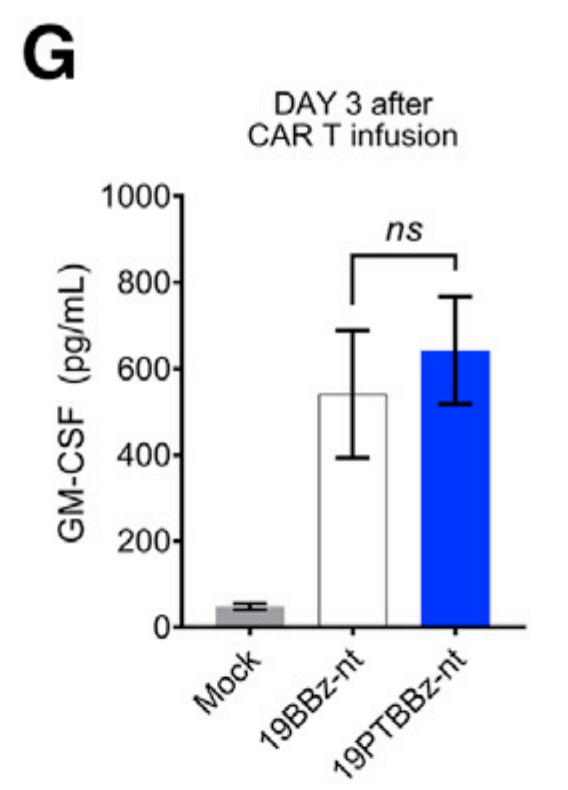
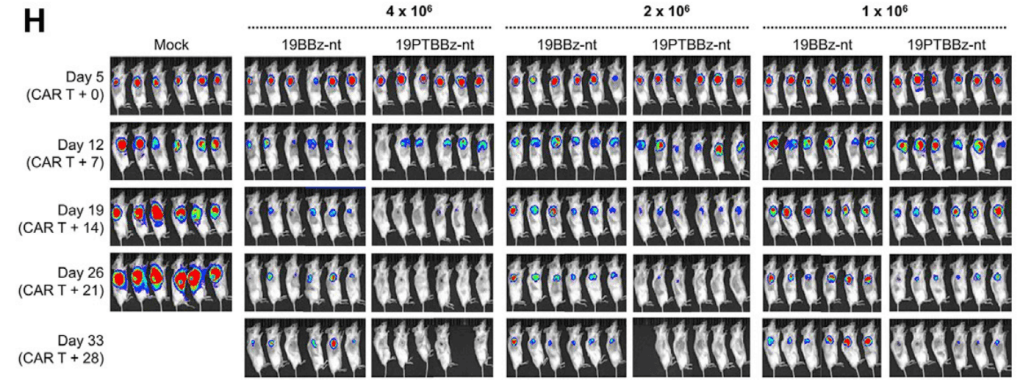

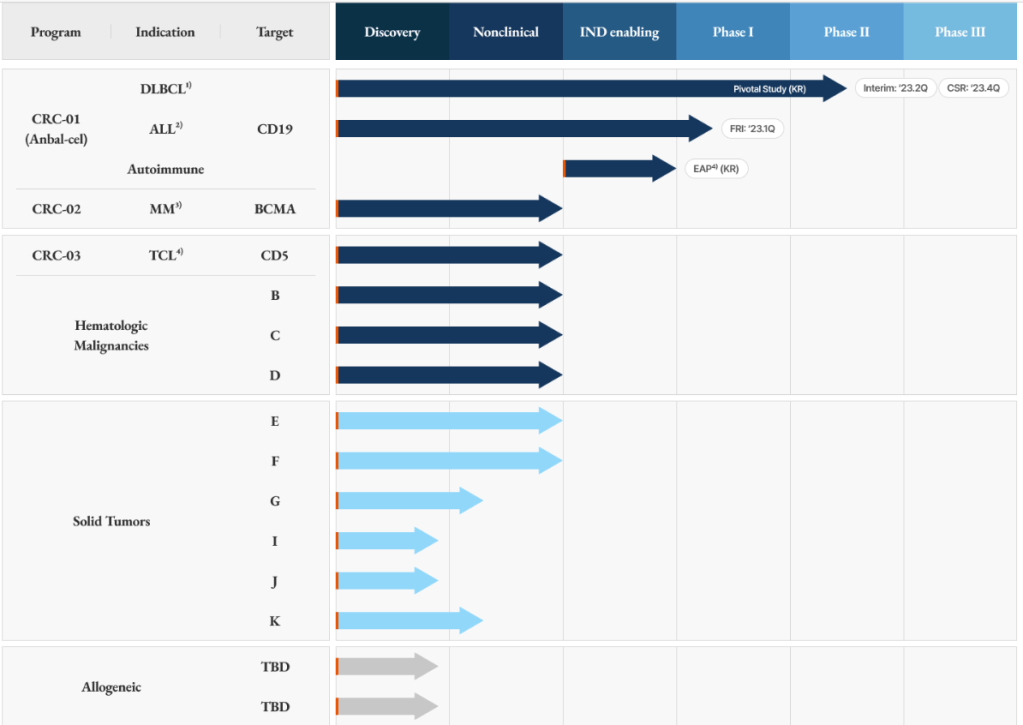
 Chugai Pharmaceutical – Eli Lilly: Orforglipron – Oral GLP-1R Agonist")
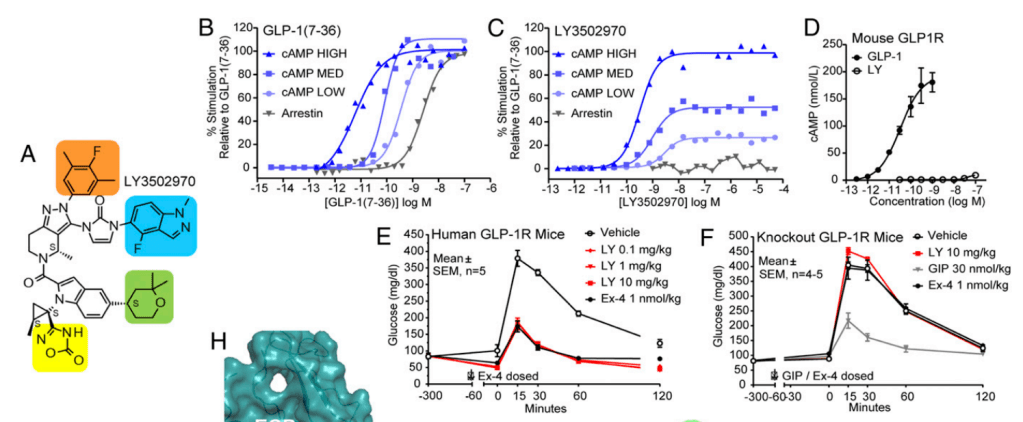
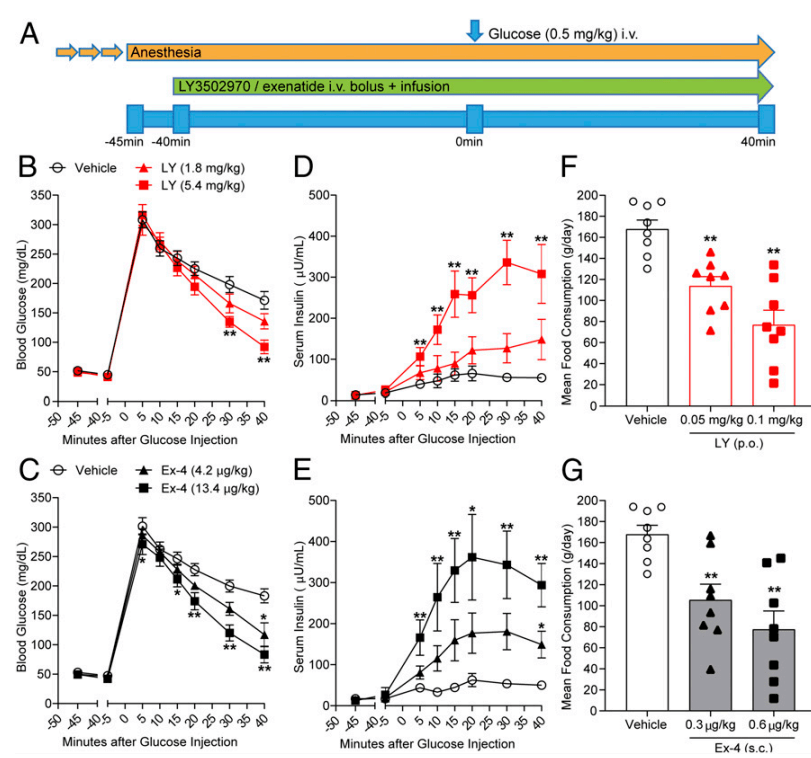
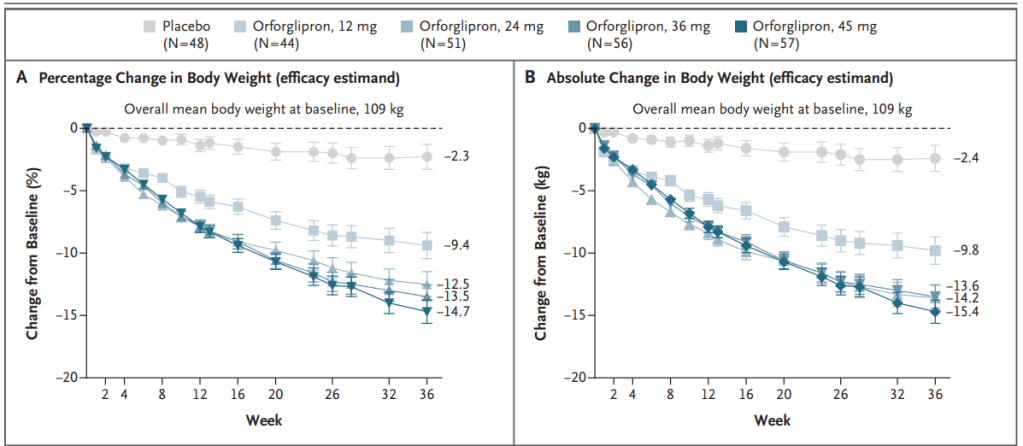
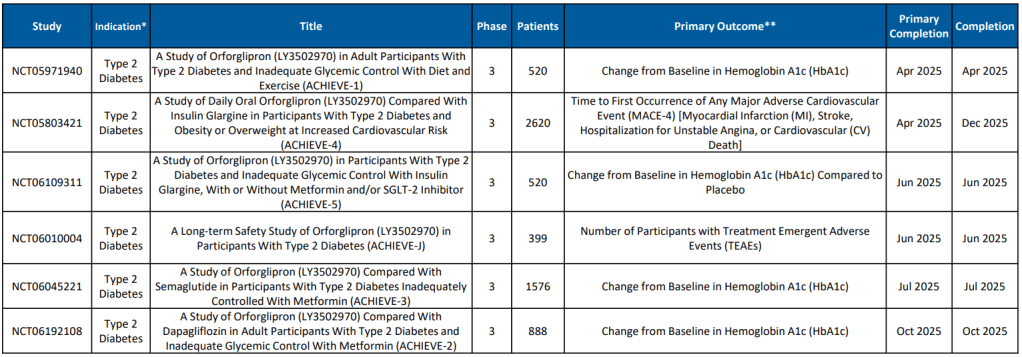
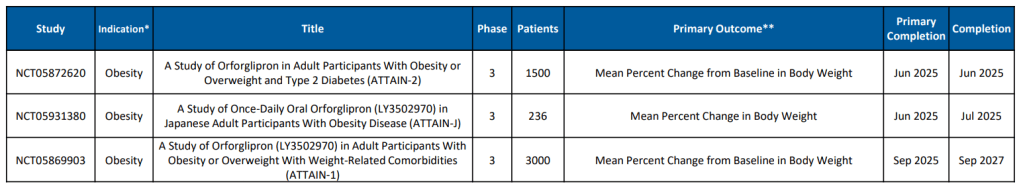
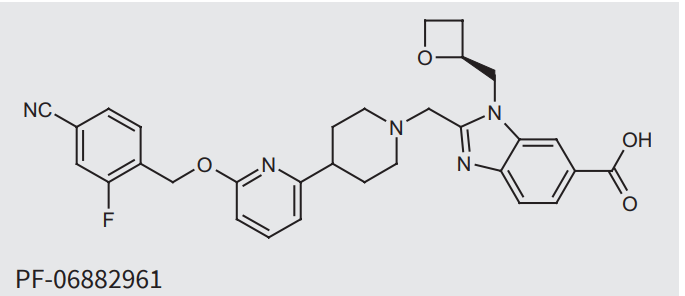
 Chugai Pharmaceutical – mRNA Display Platform & LUNA18 (pan-KRAS Inhibitor)")
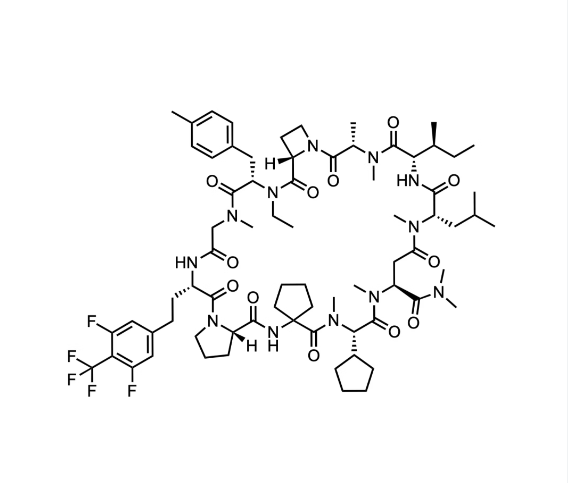
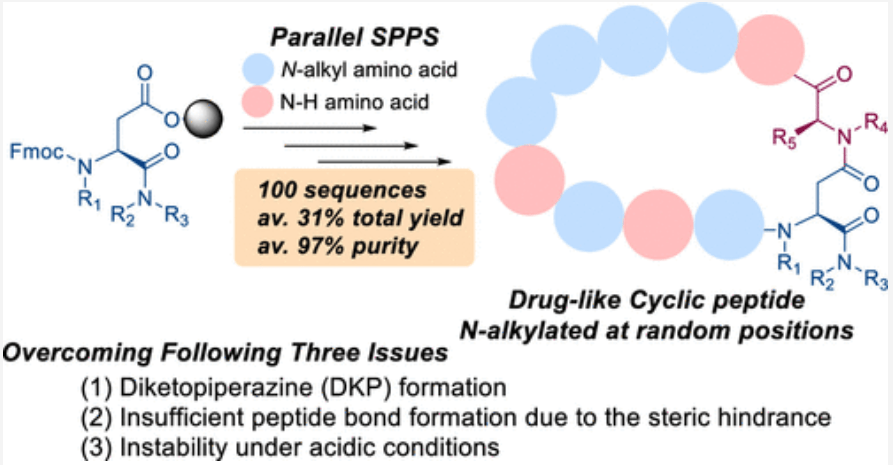
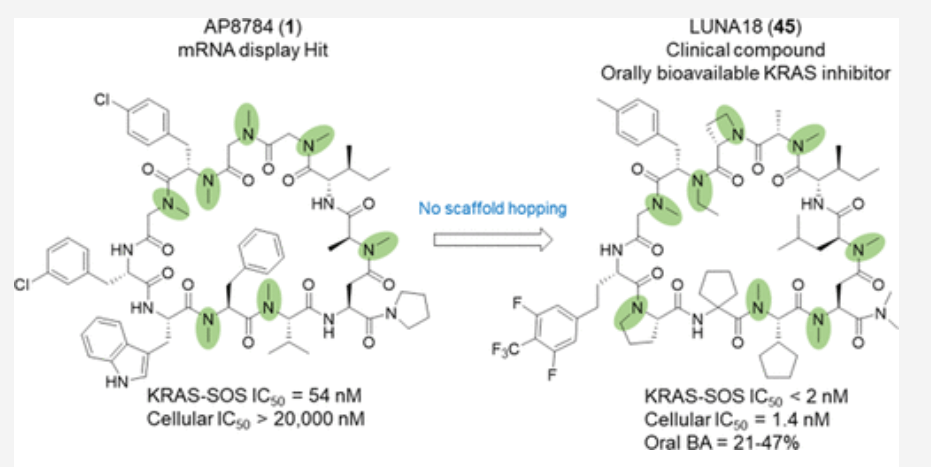

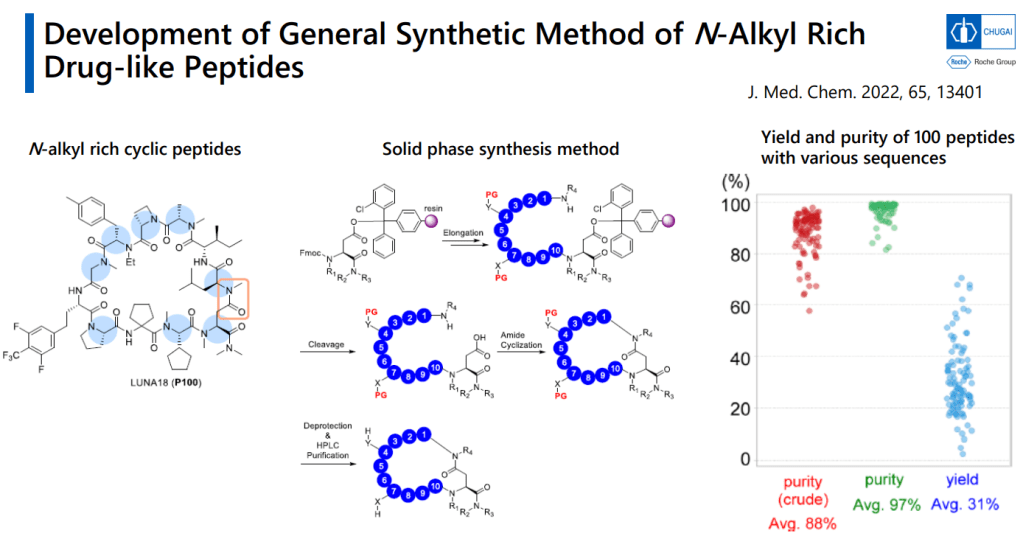
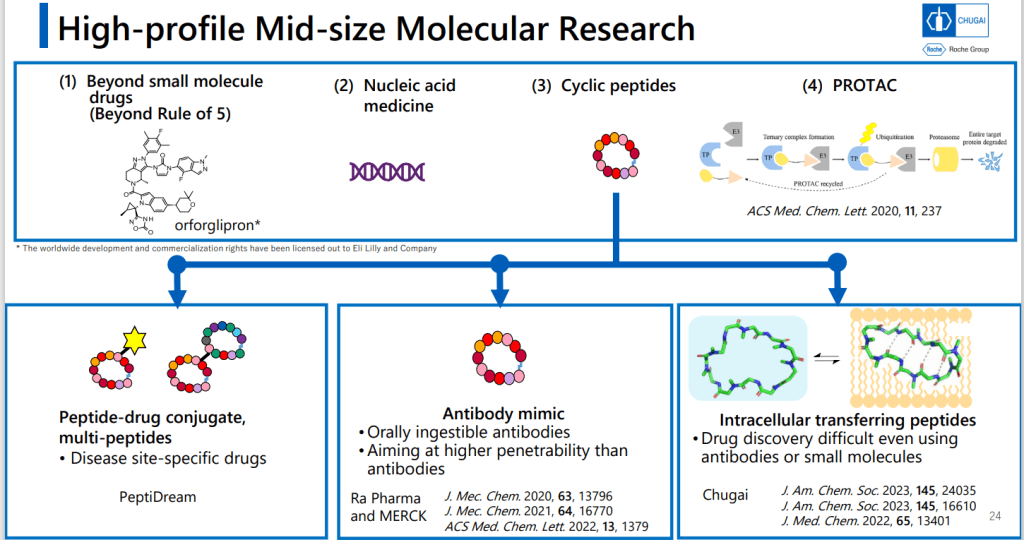
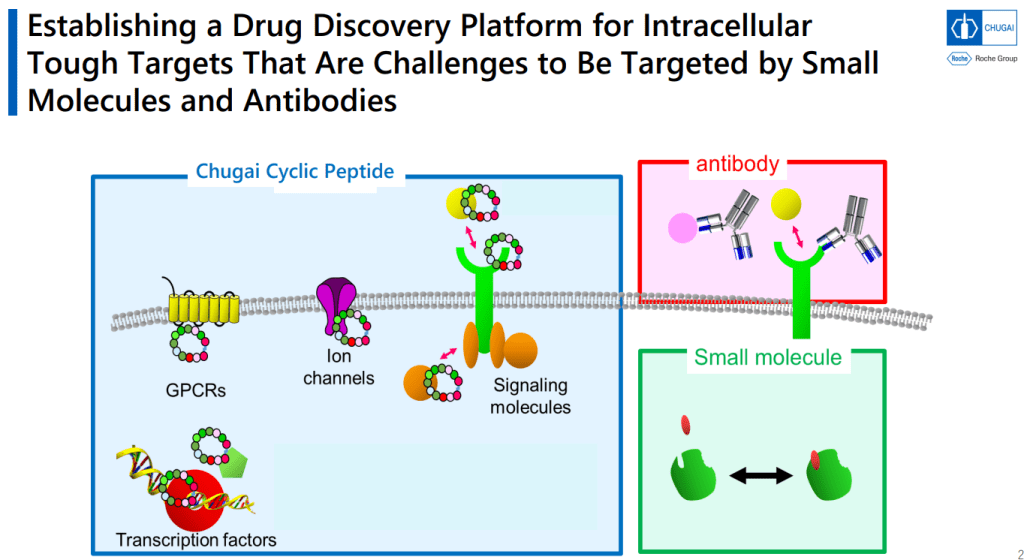
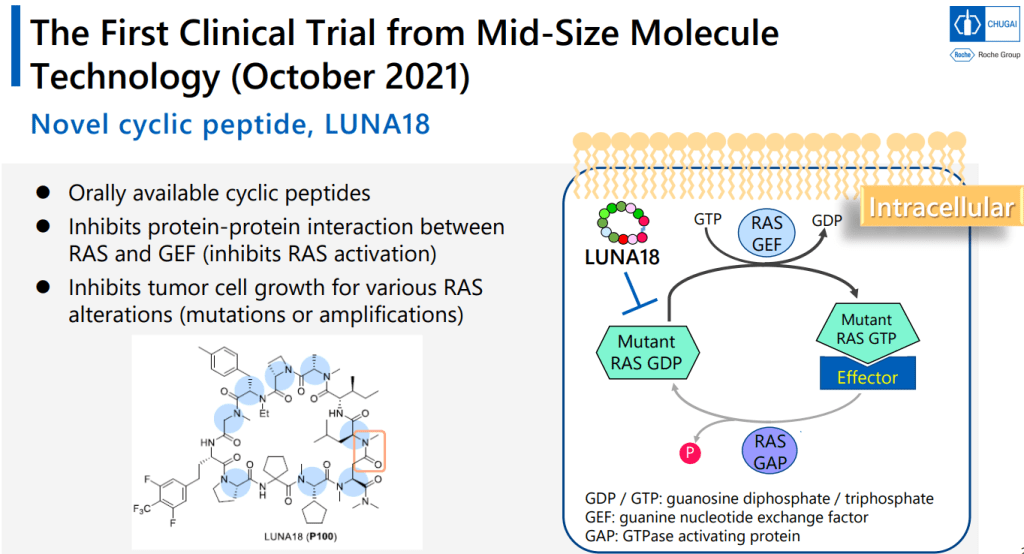
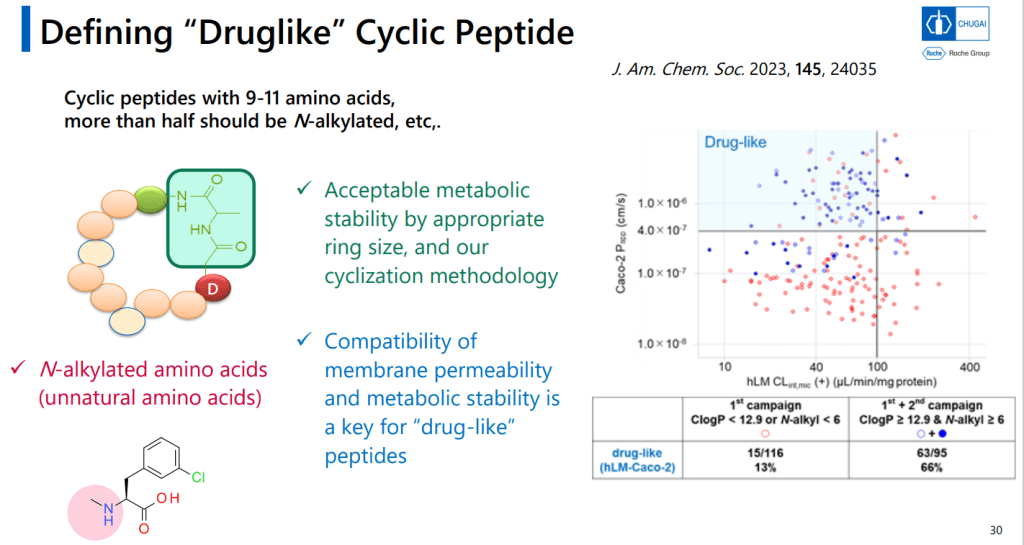
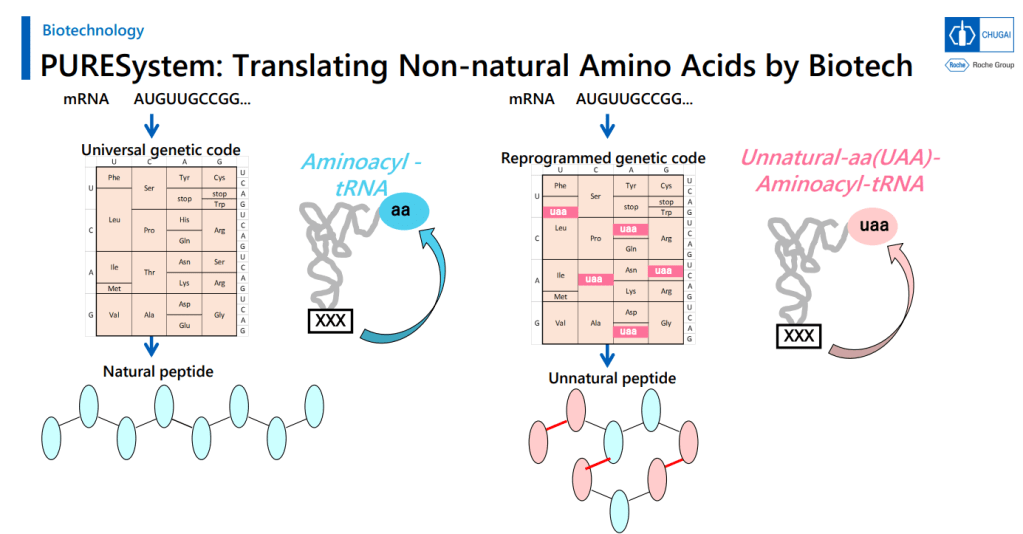
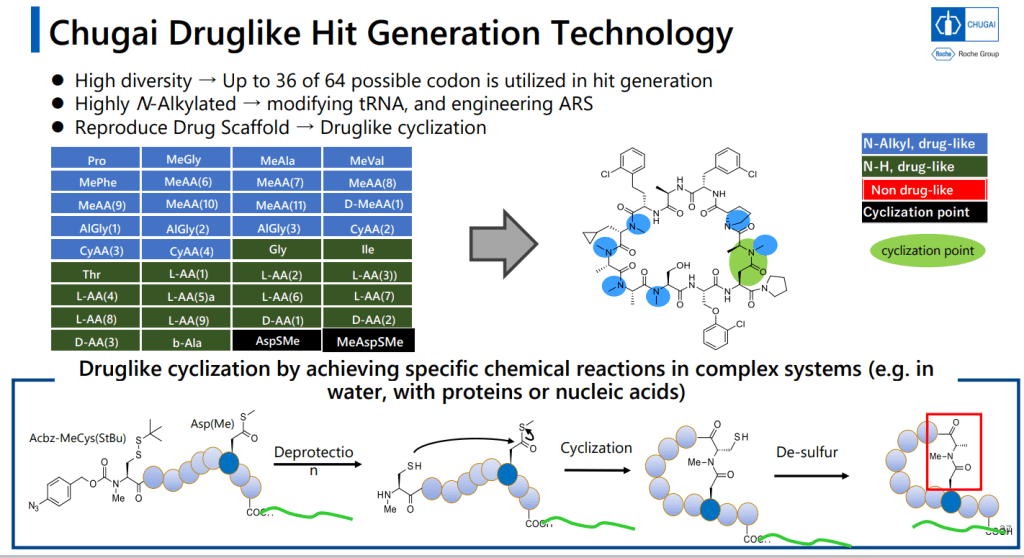
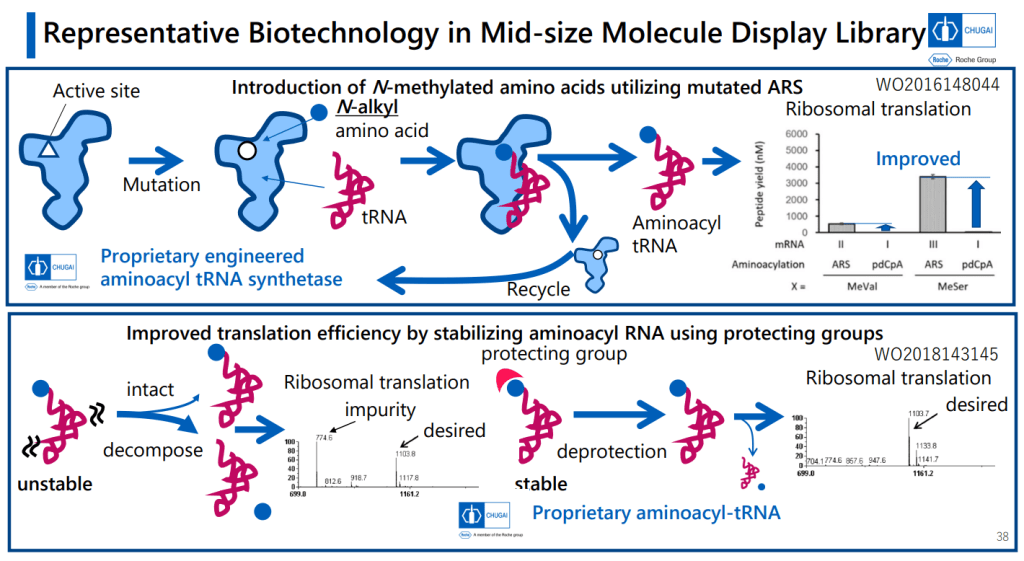
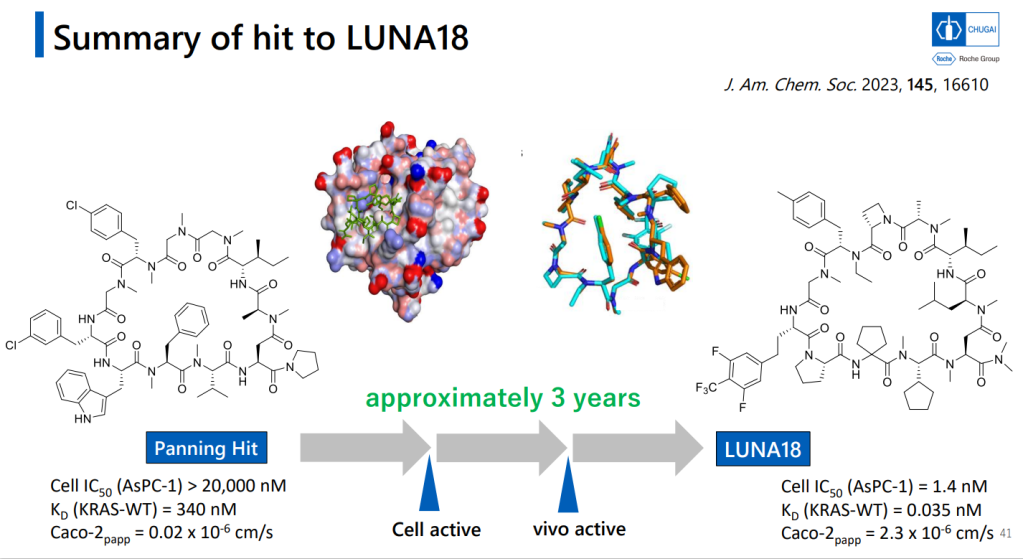
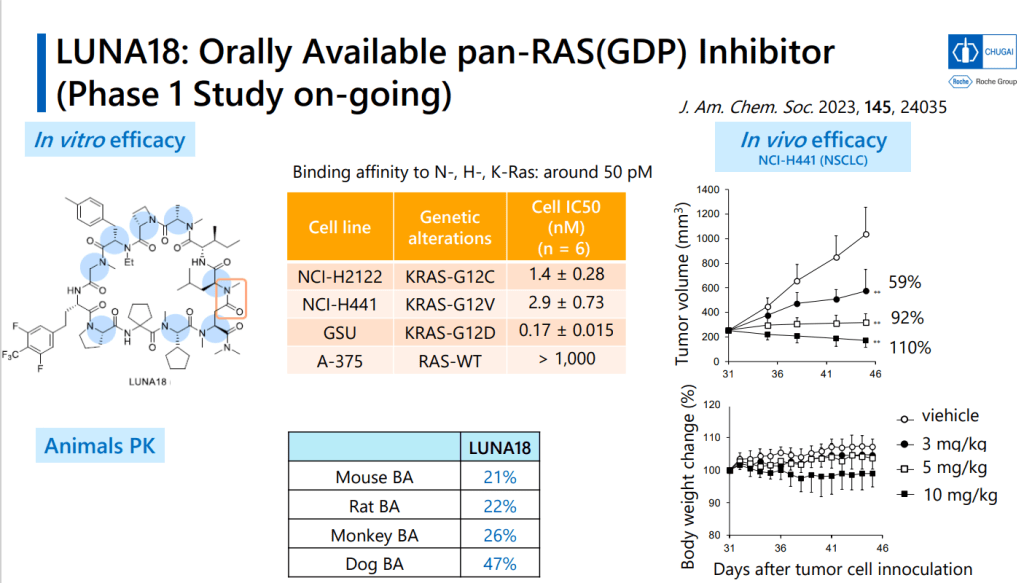
 Peptidream: Flexizyme-FIT-RaPID Platform for Macrocyclic Peptides")

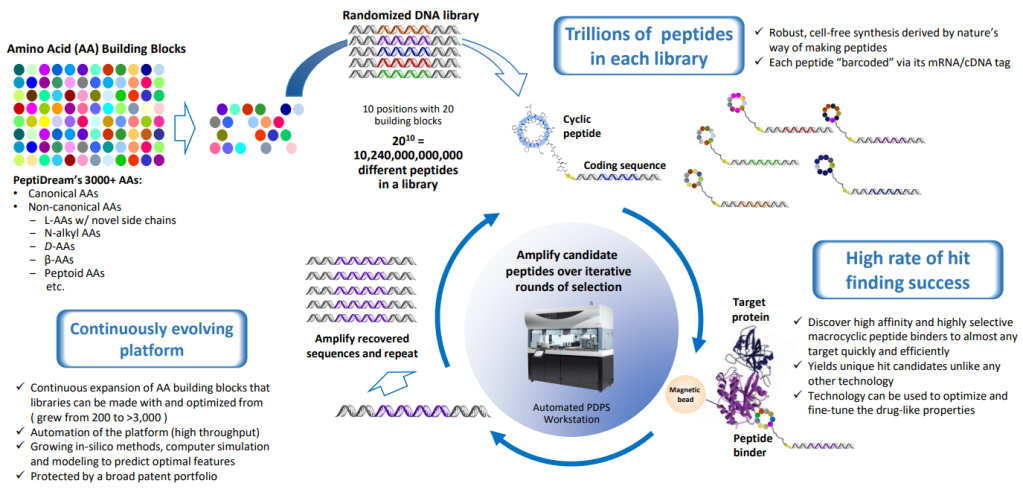


 Novo Nordisk: Semaglutide – Wegovy, Ozempic & Rybelsus for Type 2 Diabetes & Weight Loss")
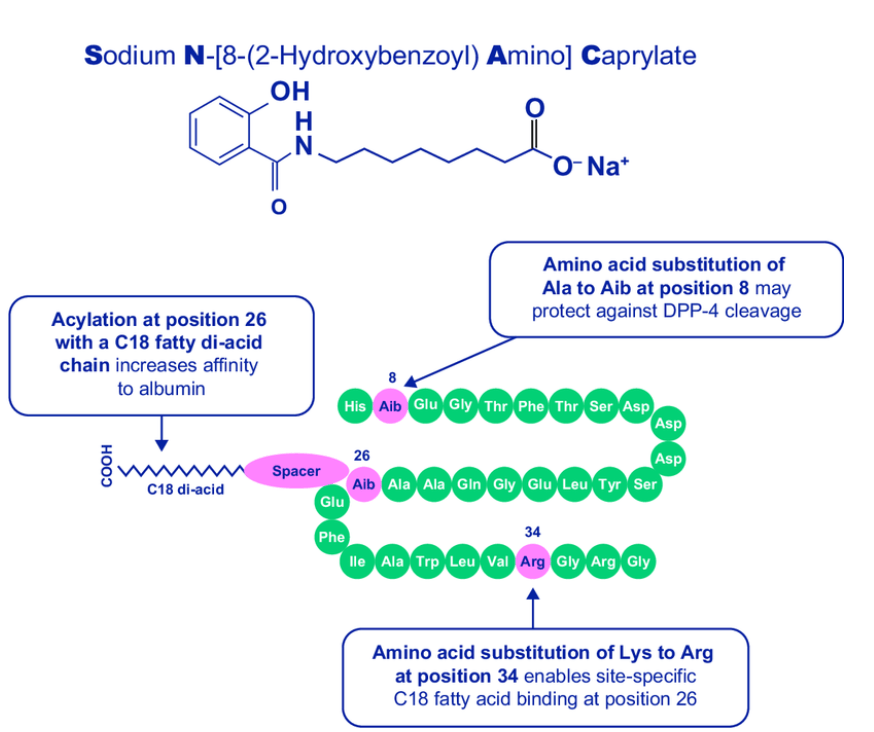
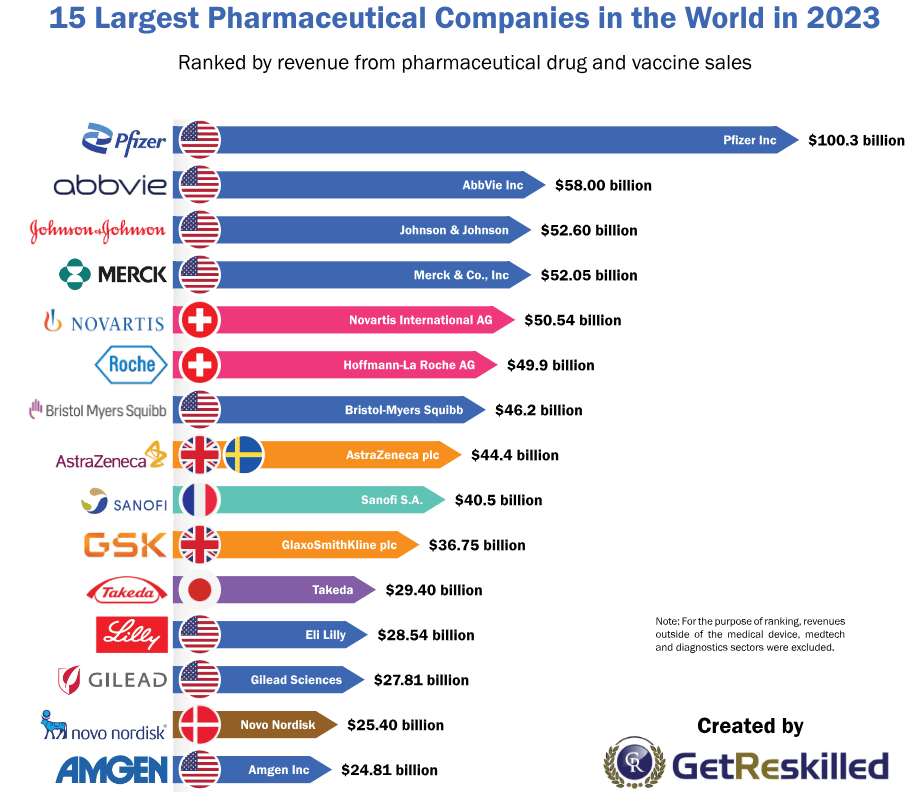


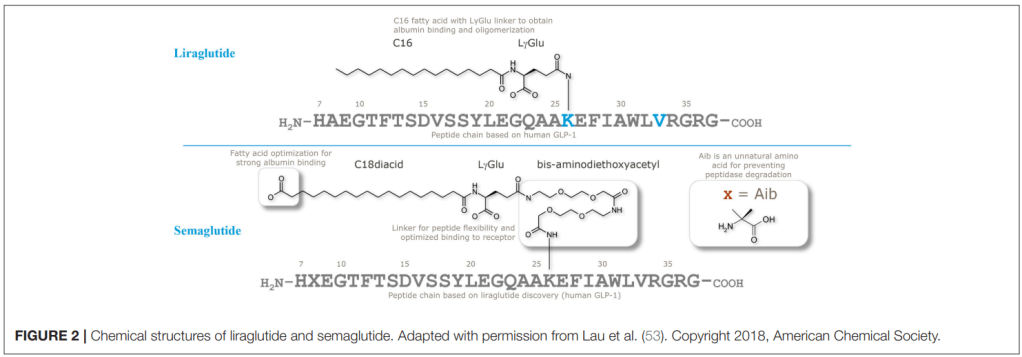
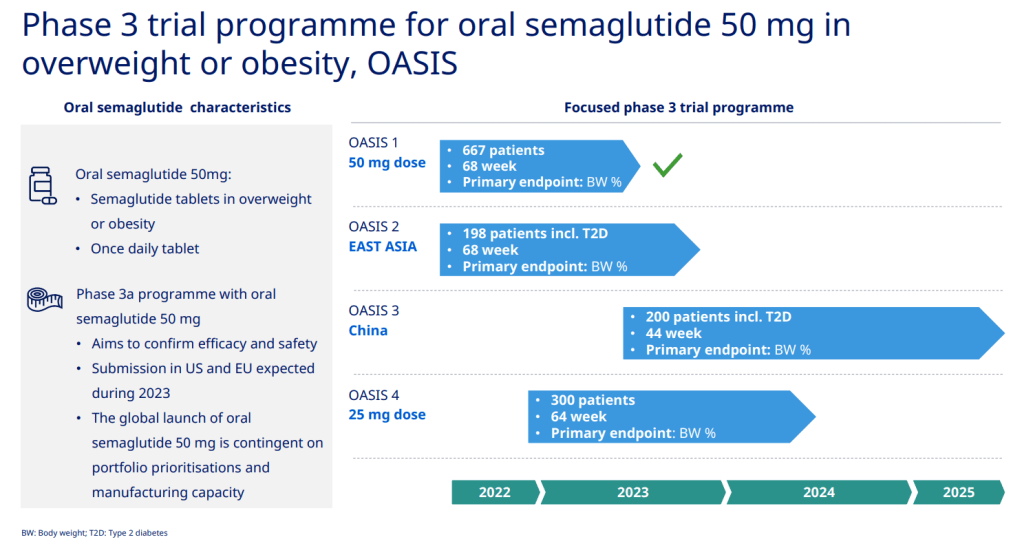
 KB금융 한국 부자 보고서와 우리금융 서울 부자 보고서에 대한 소고")


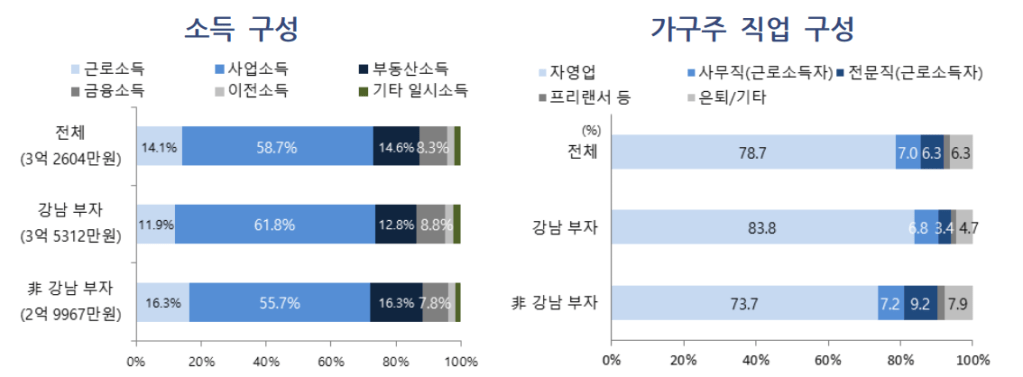
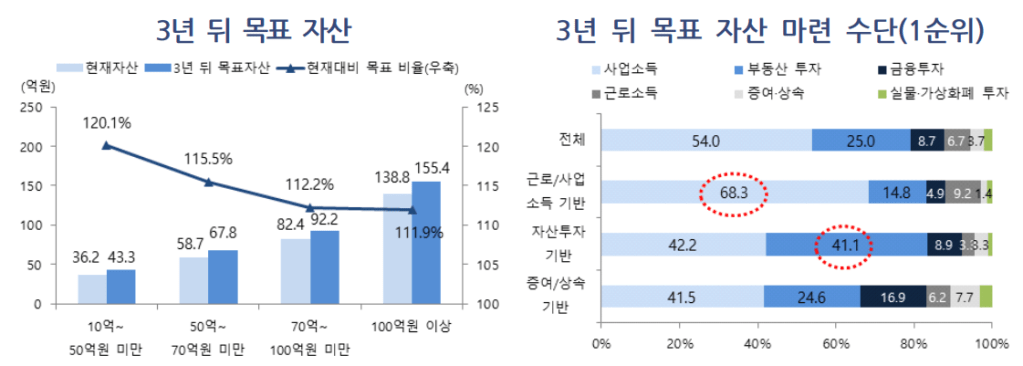
 John Crowley’s Love for Pompe Disease Drug Development from Novazyme Pharmaceuticals to Amicus Therapeutics")



 Amylyx Pharmaceuticals: ALS Drug Combo of Sodium Phenylbutyrate (PB) and Tauroursodeoxycholic Acid (TUDCA)")

 Merus NV: Multispecific T Cell Engagers")

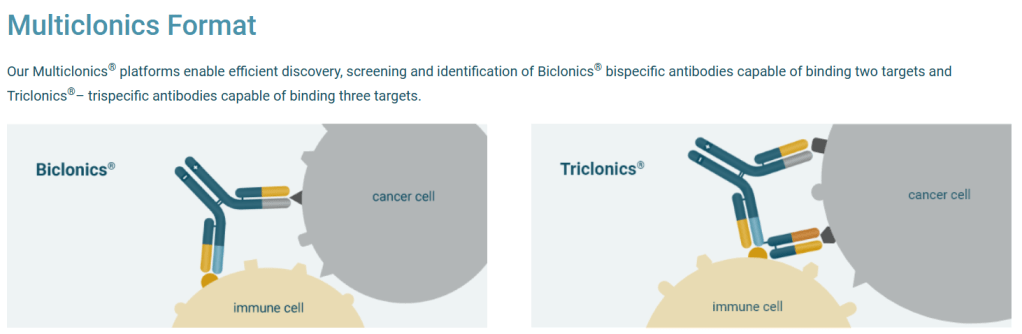

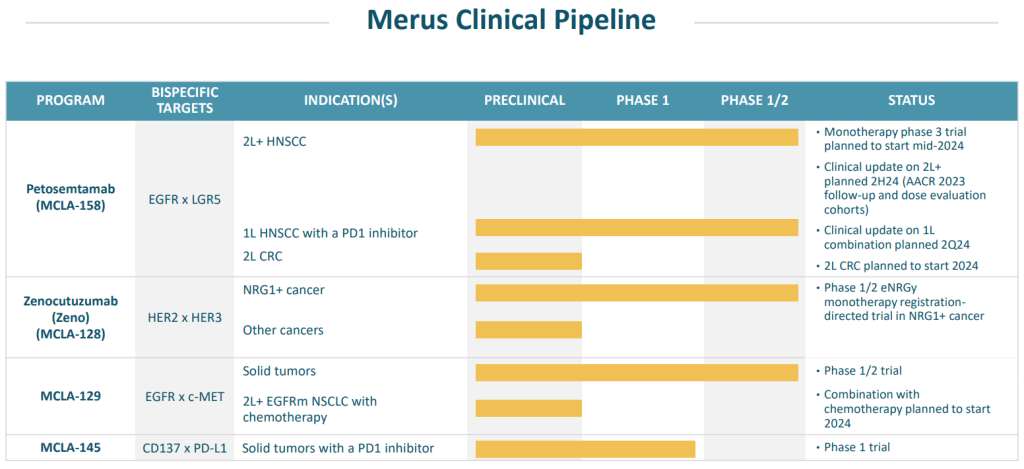
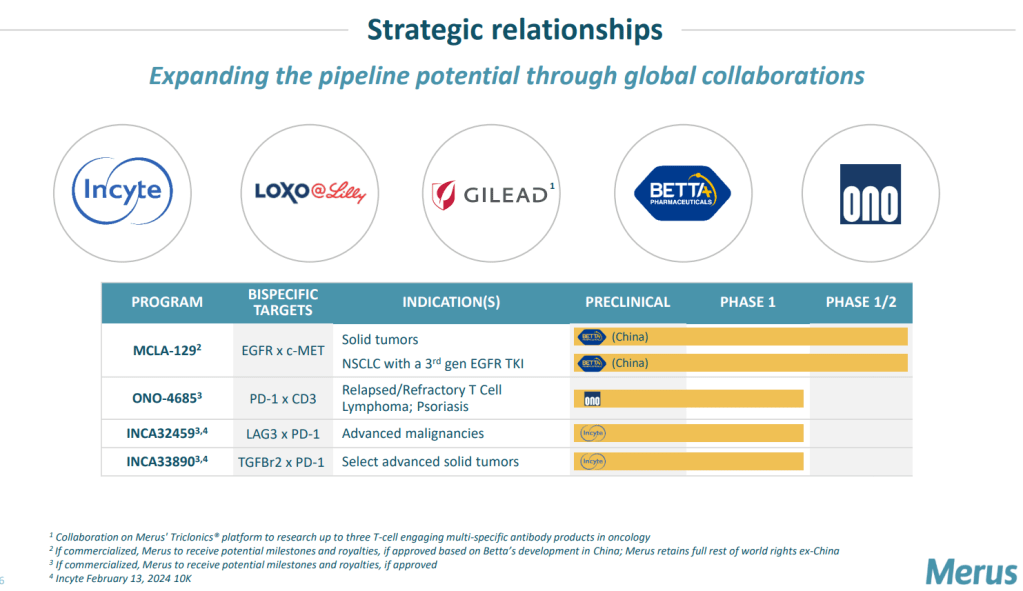
 Arvinas: PROTAC Platform Company")